Jumping library

Jumping libraries or junction-fragment libraries are collections of genomic DNA fragments generated by chromosome jumping. These libraries allow us to analyze large areas of the genome and overcome distance limitations in common cloning techniques. A jumping library clone is composed of two stretches of DNA that are usually located many kilobases away from each other. The stretch of DNA located between these two “ends” is deleted by a series of biochemical manipulations carried out at the start of this cloning technique.
Invention and early improvements
Origin
Chromosome jumping (or chromosome hopping) was first described in 1984 by Collins and Weissman.[1] At the time, cloning techniques allowed for generation of clones of limited size (up to 240kb), and cytogenetic techniques allowed for mapping such clones to a small region of a particular chromosome to a resolution of around 5-10Mb. Therefore, a major gap remained in resolution between available technologies, and no methods were available for mapping larger areas of the genome.[1]
Basic principle and original method

This technique is an extension of “chromosome walking” that allows for larger “steps” along the chromosome. If we desire to take steps of length N kb, we first require very high molecular weight DNA. Once isolated, we partially digest it with a frequent-cutting restriction enzyme (such as MboI or BamHI). Next, obtained fragments are selected for size which should be around N kb in length. DNA must then be ligated at low concentration to favour ligation into circles rather than formation of multimers. A DNA marker (such as the amber suppressor tRNA gene supF) can be included at this time point within the covalently linked circle to allow for selection of junction fragments. Circles are subsequently fully digested with a second restriction enzyme (such as EcoRI) to generate a large number of fragments. Such fragments are ligated into vectors (such as a λ vector) which should be selected for using the DNA marker introduced earlier. The remaining fragments thus represent our library of junction fragments, or “jumping library”.[1] The next step is to screen this library with a probe that represents a “starting point” of the desired “chromosome hop”, i.e. determining the location of the genome that is being interrogated. Clones obtained from this final selection step will consist of DNA that is homologous to our probe, separated by our DNA marker from another DNA sequence that was originally located N kb away (thus being called “jumping”).[1] By generating several libraries of different N values, we should eventually be able to map the entire genome and move from one location to another, while controlling direction, by any value of N desired.[1]
Early challenges and improvements
The original technique of chromosome jumping was developed in the laboratories of Collins and Weissman at Yale University in New Haven, U.S.[1] and the laboratories of Poustka and Lehrach at the European Molecular Biology Laboratory in Heidelberg, Germany.[2]
Collins and Weissman’s method[1] described above encountered some early limitations. The main concern was with avoiding non-circularized fragments. Two solutions were suggested: either screening junction fragments with a given probe or adding a second size-selection step after the ligation to separate single circular clones (monomers) from clones ligated to each other (multimers). The authors also suggested that other markers such as the λ cos site or antibiotic resistance genes should be considered (instead of the amber suppressor tRNA gene) to facilitate selection of junction clones.
Poustka and Lehrach[2] suggested that full digestion with rare-cutting restrictions enzymes (such as NotI) should be used for the first step of the library construction instead of partial digestion with a frequently cutting restriction enzyme. This would significantly reduce the number of clones from millions to thousands. However, this could create problems with circularizing the DNA fragments since these fragments would be very long, and would also lose the flexibility in choice of end points that one gets in partial digests. One suggestion for overcoming these problems would be to combine the two methods, i.e. to construct a jumping library from DNA fragments digested partially with a commonly cutting restriction enzyme and completely with a rare cutting restriction enzyme and circularizing them into plasmids cleaved with both enzymes. Several of these “combination” libraries were completed in 1986.[2][3]
In 1991, Zabarovsky et al.[4] proposed a new approach for construction of jumping libraries. This approach included the use of two separate λ vectors for library construction, and a partial filling-in reaction that removes the need for a selectable marker. This filling-in reaction worked by destroying the specific cohesive ends (resulting from restriction digests) of the DNA fragments that were nonligated and noncircularized, thus preventing them from cloning into the vectors, in a more energy-efficient and accurate manner. Furthermore, this improved technique required less DNA to start with, and also produced a library that could be transferred into a plasmid form, making it easier to store and replicate. Using this new approach, they successfully constructed a human NotI jumping library from a lymphoblastoid cell line and a human chromosome 3-specific NotI jumping library from a human chromosome 3 and mouse hybrid cell line.[4]
Current method
Second-generation or "Next-Gen" (NGS) techniques have evolved radically: the sequencing capacity has increased more than ten thousandfold and the cost has dropped by over one million fold since 2007(National Human Genome Research Institute). NGS has revolutionized the Genetic field in many ways.
Library construction
A library is often prepared by random fragmentation of DNA and ligation of common adaptor sequences.[5][6] However, the generated short reads challenge the identification of structural variants, such as indels, translocations, and duplication. Large regions of simple repeats can further complicate the alignment.[7] Alternatively, jumping library can be used with NGS for the mapping of structural variation and scaffolding of de novo assemblies.[8]
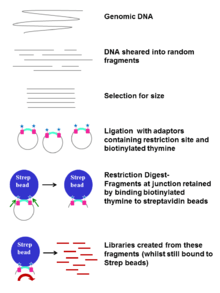
Jumping Libraries can be categorized according to the length of the incorporated DNA fragments.
- Short-jump library
3 kb genomic DNA fragments are ligated with biotinylate ends and circularized. The circular segments are then sheared into small fragments and the biotinylated fragments are selected by affinity assay for paired-end sequencing.
There are two issues related to Short-jump libraries. First, a read can pass through the biotinylated circularization junction and reduce the effective read length. Second, reads from non-jumped fragments (i.e. fragments without the circularization junction) are sequenced and reduce genomic coverage. It has been reported that non-jumped fragments range from 4% to 13%, depending on the size of selection. The first problem might be solved by shearing circles into a larger size and select for those larger fragments. The second problem can be addressed by using Custom Barcoded Jumping Library.[9][10]
- Custom Barcoded Jumping Library
This specific jumping library uses adaptors containing markers for fragment selection in combination with barcodes for multiplexing. The protocol was developed by Talkowski et al.[9] and based on mate-pair library preparation for SOLiD sequencing. The selected DNA fragment size is 3.5 – 4.5 kb. Two adaptors were involved: one containing an EcoP15I recognition site and an AC overhang; the other containing a GT overhang, a biotinylated thymine, and an oligo barcode. The circularized DNA was digested and the fragments with biotynylated adaptors were selected for(See Figure 3). The EcoP15I recognition site and barcode help to distinguish junction fragments from nonjump fragments. These targeted fragments should contain 25 to 27bp of genomic DNA, the EcoP15I recognition site, the overhang, and the barcode.[9]
- Long-jump library
This library construction process is similar to that of Short-jump library except that the condition is optimized for longer fragments(5 kb).[10]
- Fosmid-jump library
This library construction process is also similar to that of Short-jump library except that transfection using the E. coli vector is required for amplification of large (40 kb) DNA fragments. In addition, the Fosmids can be modified to facilitate the conversion into jumping library compatible with certain Next Generation Sequencers.[8][10]
Paired-end sequencing
The segments resulting from circularization during constructing jumping library are cleaved, and DNA fragments with markers will be enriched and subjected to paired-end sequencing. These DNA fragments are sequenced from both ends and generate pairs of reads. The genomic distance between the reads in each pair is approximately known and used for the assembly process. For example, a DNA clone generated by random fragmentation is about 200 bp, and a read from each end is around 180bp, overlapping each other. This should be distinguished from mate-pair sequencing, which is basically a combination of Next Generation Sequencing with jumping libraries.
Computational analysis
Different assembly tools have been developed to handle jumping library data. One example is DELLY. DELLY was developed to discover genomic structural variants and “integrates short insert paired-ends, long-range mate-pairs and split-read alignments” to detect rearrangements at sequence level.[11]
An example of joint development of new experimental design and algorithm development is demonstrated by the ALLPATHS-LG assembler.[12]
Confirmation
When used for detection of genetic and genomic changes, jumping clones require validation by Sanger sequencing.
Applications
Early applications
In the early days, chromosome walking from genetically linked DNA markers was used to identify and clone disease genes. However, the large molecular distance between known markers and the gene of interest was complicating the cloning process. In 1987, a human chromosome jumping library was constructed to clone the cystic fibrosis gene. Cystic Fibrosis is an autosomal recessive disease affecting 1 in 2000 Caucasians. This was the first disease in which the usefulness of the jumping libraries was demonstrated. Met oncogene was a marker tightly linked to the cystic fibrosis gene on human chromosome 7, and the library was screened for a jumping clone starting at this marker. The cystic fibrosis gene was determined to localize 240kb downstream of the met gene. Chromosome jumping helped reduce the mapping “steps” and bypass the highly repetitive regions in the mammalian genome.[13] Chromosome jumping also allowed the production of probes required for faster diagnosis of this and other diseases.[1]
New applications
Characterizing chromosomal rearrangements
Balanced chromosomal rearrangements can have a significant contribution to diseases, as demonstrated by the studies of leukemia.[14] However, many of them are undetected by chromosomal microarray. Karyotyping and FISH can identify balanced translocations and inversions but are labor-intensive and provide low resolution (small genomic changes are missed).
A jumping library NGS combined approach can be applied to identify such genomic changes. For example, Slade et al. applied this method to fine map a de novo balanced translocation in a child with Wilms' tumor.[15] For this study, 50 million reads were generated, but only 11.6% of these could be mapped uniquely to the reference genome, which represents approximately a sixfold coverage.
Talkowski et al.[9] compared different approaches to detect balanced chromosome alterations, and showed that modified jumping library in combination with next generation DNA sequencing is an accurate method for mapping chromosomal breakpoints. Two varieties of jumping libraries (short-jump libraries and custom barcoded jumping libraries) were tested and compared to standard sequencing libraries. For standard NGS, 200-500bp fragments are generated. About 0.03% -0.54% of fragments represent chimeric pairs, which are pairs of end-reads that are mapped to two different chromosomes. Therefore, very few fragments cover the breakpoint area. When using short-jump libraries with fragments of 3.2–3.8kb, the percentage of chimeric pairs increased to 1.3%. With Custom Barcoded Jumping Libraries, the percentage of chimeric pairs further increased to 1.49%.[9]
Prenatal diagnosis
Conventional cytogenetic testing cannot offer the gene-level resolution required to predict the outcome of a pregnancy and whole genome deep sequencing is not practical for routine prenatal diagnosis. Whole-genome jumping library could complement conventional prenatal testing. This novel method was successfully applied to idenfity a case of CHARGE syndrome.[6]
De novo assembly
In metagenomics, regions of the genomes that are shared between strains are typically longer than the reads. This complicates the assembly process and makes reconstructing individual genomes for a species a daunting task.[10] Chimeric pairs that are mapped far apart in the genome can facilitate the de novo assembly process. By using a longer-jump library, Ribeiro et al. demonstrated that the assemblies of bacterial genomes were of high quality while reducing both cost and time.[16]
Limitation
The cost of sequencing has dropped dramatically during the past few years while the cost of construction of jumping libraries has not. Therefore,as newsequencing technologies and bioinformatic tools are developed, jumping libraries may become redundant.
External links
- DELLY: Structural variant discovery by integrated paired-end and split-read analysis
- ALLPATHS-LG: de novo assembly of whole-genome shotgun microreads[17]
See also
References
- 1 2 3 4 5 6 7 8 Collins, FS; Weissman, SM (November 1984). "Directional cloning of DNA fragments at a large distance from an initial probe: a circularization method.". Proceedings of the National Academy of Sciences of the United States of America. 81 (21): 6812–6. doi:10.1073/pnas.81.21.6812. PMID 6093122.
- 1 2 3 Poustka, Annemarie; Lehrach, Hans (1 January 1986). "Jumping libraries and linking libraries: the next generation of molecular tools in mammalian genetics". Trends in Genetics. 2: 174–179. doi:10.1016/0168-9525(86)90219-2.
- ↑ Poustka, A; Pohl, TM; Barlow, DP; Frischauf, AM; Lehrach, H (Jan 22–28, 1987). "Construction and use of human chromosome jumping libraries from NotI-digested DNA.". Nature. 325 (6102): 353–5. doi:10.1038/325353a0. PMID 3027567.
- 1 2 Zabarovsky, ER; Boldog, F; Erlandsson, R; Kashuba, VI; Allikmets, RL; Marcsek, Z; Kisselev, LL; Stanbridge, E; Klein, G; Sumegi, J (December 1991). "New strategy for mapping the human genome based on a novel procedure for construction of jumping libraries.". Genomics. 11 (4): 1030–9. doi:10.1016/0888-7543(91)90029-e. PMID 1783374.
- ↑ Shendure, J; Ji, H (October 2008). "Next-generation DNA sequencing.". Nature Biotechnology. 26 (10): 1135–45. doi:10.1038/nbt1486. PMID 18846087.
- 1 2 Talkowski, ME; Ordulu, Z; Pillalamarri, V; Benson, CB; Blumenthal, I; Connolly, S; Hanscom, C; Hussain, N; Pereira, S; Picker, J; Rosenfeld, JA; Shaffer, LG; Wilkins-Haug, LE; Gusella, JF; Morton, CC (Dec 6, 2012). "Clinical diagnosis by whole-genome sequencing of a prenatal sample.". The New England Journal of Medicine. 367 (23): 2226–32. doi:10.1056/NEJMoa1208594. PMID 23215558.
- ↑ Meldrum, C; Doyle, MA; Tothill, RW (November 2011). "Next-generation sequencing for cancer diagnostics: a practical perspective.". The Clinical biochemist. Reviews / Australian Association of Clinical Biochemists. 32 (4): 177–95. PMID 22147957.
- 1 2 Williams, L. J. S.; Tabbaa, D. G.; Li, N.; Berlin, A. M.; Shea, T. P.; MacCallum, I.; Lawrence, M. S.; Drier, Y.; Getz, G.; Young, S. K.; Jaffe, D. B.; Nusbaum, C.; Gnirke, A. (16 July 2012). "Paired-end sequencing of Fosmid libraries by Illumina". Genome Research. 22 (11): 2241–2249. doi:10.1101/gr.138925.112.
- 1 2 3 4 5 Talkowski, ME; Ernst, C; Heilbut, A; Chiang, C; Hanscom, C; Lindgren, A; Kirby, A; Liu, S; Muddukrishna, B; Ohsumi, TK; Shen, Y; Borowsky, MZ; Daly, MJ; Morton, CC; Gusella, JF (Apr 8, 2011). "Next-generation sequencing strategies enable routine detection of balanced chromosome rearrangements for clinical diagnostics and genetic research.". American Journal of Human Genetics. 88 (4): 469–81. doi:10.1016/j.ajhg.2011.03.013. PMID 21473983.
- 1 2 3 4 Nagarajan, Niranjan; Pop, Mihai (29 January 2013). "Sequence assembly demystified". Nature Reviews Genetics. 14 (3): 157–167. doi:10.1038/nrg3367.
- ↑ Rausch, T.; Zichner, T.; Schlattl, A.; Stutz, A. M.; Benes, V.; Korbel, J. O. (7 September 2012). "DELLY: structural variant discovery by integrated paired-end and split-read analysis". Bioinformatics. 28 (18): i333–i339. doi:10.1093/bioinformatics/bts378. PMC 3436805
 . PMID 22962449.
. PMID 22962449. - ↑ Gnerre, S; Maccallum, I; Przybylski, D; Ribeiro, FJ; Burton, JN; Walker, BJ; Sharpe, T; Hall, G; Shea, TP; Sykes, S; Berlin, AM; Aird, D; Costello, M; Daza, R; Williams, L; Nicol, R; Gnirke, A; Nusbaum, C; Lander, ES; Jaffe, DB (Jan 25, 2011). "High-quality draft assemblies of mammalian genomes from massively parallel sequence data.". Proceedings of the National Academy of Sciences of the United States of America. 108 (4): 1513–8. doi:10.1073/pnas.1017351108. PMID 21187386.
- ↑ Rommens, J.; Iannuzzi, M.; Kerem, B; Drumm, M.; Melmer, G; Dean, M; Rozmahel, R; Cole, J.; Kennedy, D; Hidaka, N; et al. (8 September 1989). "Identification of the cystic fibrosis gene: chromosome walking and jumping". Science. 245 (4922): 1059–1065. doi:10.1126/science.2772657. PMID 2772657.
- ↑ Rowley, J.D. (1979). "chromosome abnormalities in leukemia". Blood Transfus.
- ↑ Slade, I.; Stephens, P.; Douglas, J.; Barker, K.; Stebbings, L.; Abbaszadeh, F.; Pritchard-Jones, K.; Cole, R.; Pizer, B.; Stiller, C.; Vujanic, G.; Scott, R. H.; Stratton, M. R.; Rahman, N. (30 November 2009). "Constitutional translocation breakpoint mapping by genome-wide paired-end sequencing identifies HACE1 as a putative Wilms tumour susceptibility gene". Journal of Medical Genetics. 47 (5): 342–347. doi:10.1136/jmg.2009.072983.
- ↑ Ribeiro, F. J.; Przybylski, D.; Yin, S.; Sharpe, T.; Gnerre, S.; Abouelleil, A.; Berlin, A. M.; Montmayeur, A.; Shea, T. P.; Walker, B. J.; Young, S. K.; Russ, C.; Nusbaum, C.; MacCallum, I.; Jaffe, D. B. (24 July 2012). "Finished bacterial genomes from shotgun sequence data". Genome Research. 22 (11): 2270–2277. doi:10.1101/gr.141515.112.
- ↑ Butler, J; MacCallum, I; Kleber, M; Shlyakhter, IA; Belmonte, MK; Lander, ES; Nusbaum, C; Jaffe, DB (May 2008). "ALLPATHS: de novo assembly of whole-genome shotgun microreads.". Genome Research. 18 (5): 810–20. doi:10.1101/gr.7337908. PMID 18340039.