Electrode potential
Electrode potential, E, in chemistry or electrochemistry, according to a IUPAC definition,[1] is the electromotive force of a cell built of two electrodes:
- on the left-hand side is the standard hydrogen electrode, and
- on the right-hand side is the electrode the potential of which is being defined.
By convention:
- ECell = ECathode − EAnode
From the above, for the cell with the standard hydrogen electrode (potential of 0 by convention), one obtains:
- ECell = ERight − 0 = EElectrode
The left-right convention is consistent with the international agreement that redox potentials be given for reactions written in the form of reduction half-reactions.
Electrode potential is measured in volts (V).
Origin and interpretation
Electrode potential appears at the interface between an electrode and electrolyte due to the transfer of charged species across the interface, specific adsorption of ions at the interface, and specific adsorption/orientation of polar molecules, including those of the solvent.
Electrode potential is the electric potential on an electrode component. In a cell, there will be an electrode potential for the cathode, and an electrode potential for the anode. The difference between the two electrode potentials equals the cell potential.
ECell = ECathode − EAnode
The measured electrode potential may be either that at equilibrium on the working electrode ("reversible potential"), or a potential with a non-zero net reaction on the working electrode but zero net current ("corrosion potential", "mixed potential"), or a potential with a non-zero net current on the working electrode (like in galvanic corrosion or voltammetry). Reversible potentials can be sometimes converted to the standard electrode potential for a given electroactive species by extrapolation of the measured values to the standard state.
The value of the electrode potential under non-equilibrium depends on the nature and composition of the contacting phases, and on the kinetics of electrode reactions at the interface (see Butler-Volmer equation).
An operational assumption for determinations of the electrode potentials with the standard hydrogen electrode involves this reference electrode with hydrogen ion in an ideal solution having is "zero potential at all temperatures" equivalently to standard enthalpy of formation of hydrogen ion is also "zero at all temperatures".
Measurement
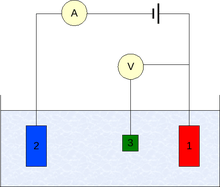
The measurement is generally conducted using a three-electrode setup (see the drawing):
- Working electrode
- Counter electrode
- Reference electrode (standard hydrogen electrode or an equivalent)
In case of non-zero net current on the electrode, it is essential to minimize the ohmic IR-drop in the electrolyte, e.g., by positioning the reference electrode near the surface of the working electrode (e.g., see Luggin capillary), or by using a supporting electrolyte of sufficiently high conductivity. The potential measurements are performed with the positive terminal of the electrometer connected to working electrode and the negative terminal to the reference electrode.
Sign conventions
Historically, two conventions for sign for the electrode potential have formed:[2]
- Convention "Nernst-Lewis-Latimer" (sometimes referred to as "American");
- Convention "Gibbs-Ostwald-Stockholm" (sometimes referred to as "European").
In 1953 in Stockholm[3] IUPAC recognized that either of the conventions is permissible; however, it unanimously recommended that only the magnitude expressed according to the convention (2) be called "the electrode potential". To avoid possible ambiguities, the electrode potential thus defined can also be referred to as Gibbs-Stockholm electrode potential.
In both conventions, the EMF of the half-cell is referred to the standard hydrogen electrode as the zero point.
According to the Nernst-Lewis-Latimer convention (1), the sign of the potential is determined by the change of the sign of the standard Gibbs free energy of the half-cell reaction. This reaction transfers nF of the positive electric charge across the phase boundary from the left to the right of the half-cell, as shown in the half-cell diagram. The relation is written as:

where: ΔG - the standard Gibbs energy for the half-cell reaction, n - number of electrons transferred across the across the phase boundary in the above reaction, F - the Faraday constant, E - the half-cell potential according to the Nernst-Lewis-Latimer convention.
Therefore, according to the convention (1), the value of EMF is bivariant (with two variants), i.e., reversing the electrode reaction, and reversing the corresponding cell diagram, will cause the change of sign of ΔG and E, even though the polarity of the cell measured with a direct-current voltmeter does not necessarily reverse. This convention has been historically (sometimes still is) used to express anodic oxidation potentials or cathodic reduction potentials of half-cells. The reduction potentials expressed according to (1) have the same sign as those expressed according to the convention (2).
According to the Gibbs-Stockholm convention (2), the sign of the electrode potential is determined by the polarity of the cell terminal measured against the standard hydrogen electrode using an instrument for DC-potential measurements, when this circuit is closed and balanced (i.e., the current on the measurement circuit is nil). In convention (2), the value of the measured EMF is invariant of the direction of the infinitesimally small changes of the electric current that may be allowed on the measured electrode and render it either cathodic or anodic. Therefore, the value of E does not change with the direction of the (very small) current, even though ΔG of the corresponding reaction is bivariant (can be of either positive or negative sign).
Potential difference of a cell assembled of two electrodes
Potential of a cell assembled of two electrodes can be determined from the two individual electrode potentials using:
ΔVcell = Ered,cathode - Ered,anode
or, equivalently,
ΔVcell = Ered,cathode + Eoxy,anode
This follows from the IUPAC definition of the electric potential difference of a galvanic cell,[4] according to which the electric potential difference of a cell is the difference of the potentials of the electrodes on the right and the left of the galvanic cell. When ΔVcell is positive, then positive electrical charge flows through the cell from the left electrode (anode) to the right electrode (cathode).
See also
- Standard electrode potential
- Absolute electrode potential
- Table of standard electrode potentials
- Electric potential
- Galvani potential
- Volta potential
- Potential difference (voltage)
- Overpotential
- Thermodynamic activity
- Nernst equation
References
- ↑ IUPAC. Compendium of Chemical Terminology, 2nd ed. (the "Gold Book"). Compiled by A. D. McNaught and A. Wilkinson. Blackwell Scientific Publications, Oxford (1997). XML on-line corrected version: http://goldbook.iupac.org (2006- ) created by M. Nic, J. Jirat, B. Kosata; updates compiled by A. Jenkins. ISBN 0-9678550-9-8. doi:10.1351/goldbook. Entry: "Electrode Potential"
- ↑ C.A. Hamel, "The Encyclopedia of Electrochemistry", Reinhold Publishing Corporation, New York-Chapman & Hall Ltd., London, 1964, s. 429-431.
- ↑ P. van Rysselberghe, "Bericht der Kommission für elektrochemische Nomenklatur und Definitionen", Z. Electrochem., 58 (1954), 530-535.
- ↑ IUPAC Gold Book. Definition of the potential difference of a galvanic cell. http://goldbook.iupac.org/E01934.html