Biotinidase deficiency
| Biotinidase deficiency | |
|---|---|
|
| |
| Classification and external resources | |
| Specialty | endocrinology |
| ICD-10 | E53.8 |
| ICD-9-CM | 277.6 |
| OMIM | 253260 |
| DiseasesDB | 29822 |
| eMedicine | ped/239 |
| MeSH | D028921 |
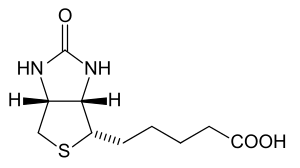
Biotinidase deficiency is an autosomal recessive metabolic disorder in which biotin is not released from proteins in the diet during digestion or from normal protein turnover in the cell. This situation results in biotin deficiency.
Biotin, also called vitamin B7, is an important water-soluble nutrient that aids in the metabolism of fats, carbohydrates, and proteins. Biotin deficiency can result in behavioral disorders, lack of coordination, learning disabilities and seizures. Biotin supplementation can alleviate and sometimes totally arrest such symptoms.
Symptoms
Symptoms of a biotinidase deficiency can appear several days after birth. These include: seizures, hypotonia and muscle/limb weakness, ataxia, paresis, hearing loss, optic atrophy, skin rashes (including seborrheic dermatitis and psoriasis), and alopecia. If left untreated, the disorder can rapidly lead to coma and death.
Biotinidase deficiency can also appear later in life. This is referred to as "late-onset" biotinidase deficiency. The symptoms are similar, but perhaps more mild, because if an individual survives the neonatal period they likely have some residual activity of biotin-related enzymes. Studies[1][2] have noted individuals who were asymptomatic until adolescence or early adulthood. One study pointed out that untreated individuals may not show symptoms until age 21.[3] Furthermore, in rare cases, even individuals with profound deficiencies of biotinidase can be asymptomatic.[1]
Symptom severity is predictably correlated with the severity of the enzyme defect. Profound biotinidase deficiency refers to situations where enzyme activity is 10% or less.[2] Individuals with partial biotinidase deficiency may have enzyme activity of 10-30%.[2]
Functionally, there is no significant difference between dietary biotin deficiency and genetic loss of biotin-related enzyme activity. In both cases, supplementation with biotin can often restore normal metabolic function and proper catabolism of leucine and isoleucine.
The symptoms of biotinidase deficiency (and dietary deficiency of biotin) can be quite severe. A 2004 case study from Metametrix [4] detailed the effects of biotin deficiency, including aggression, cognitive delay, and reduced immune function.
Genetics
Mutations in the BTD gene cause biotinidase deficiency. Biotinidase is the enzyme that is made by the BTD gene. Many mutations that cause the enzyme to be nonfunctional or to be produced at extremely low levels have been identified. Biotin is a vitamin that is chemically bound to proteins. (Most vitamins are only loosely associated with proteins.) Without biotinidase activity, the vitamin biotin cannot be separated from foods and therefore cannot be used by the body. Another function of the biotinidase enzyme is to recycle biotin from enzymes that are important in metabolism (processing of substances in cells). When biotin is lacking, specific enzymes called carboxylases cannot process certain proteins, fats, or carbohydrates. Specifically, two essential branched-chain amino acids (leucine and isoleucine) are metabolized differently.
Individuals lacking functional biotinidase enzymes can still have normal carboxylase activity if they ingest adequate amounts of biotin. The standard treatment regimen calls for 5–10 mg of biotin per day.[5]
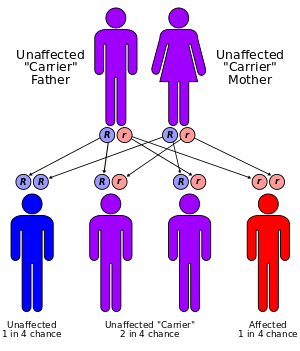
Biotinidase deficiency is inherited in an autosomal recessive pattern, which means the defective gene is located on an autosome, and two copies of the defective gene - one from each parent - must be inherited for a person to be affected by the disorder. The parents of a child with an autosomal recessive disorder are usually not affected by the disorder, but are carriers of one copy of the defective gene. If both parents are carriers for the biotinidase deficiency, there is a 25% chance that their child will be born with it, a 50% chance the child will be a carrier, and a 25% chance the child will be unaffected.
The chromosomal locus is at 3p25. The BTD gene has 4 exons of lengths 79 bp, 265 bp, 150 bp and 1502 bp, respectively. There are at least 21 different mutations that have been found to lead to biotinidase deficiency. The most common mutatations in severe biotinidase deficiency (<10% normal enzyme activity) are: p.Cys33PhefsX36, p.Gln456His, p.Arg538Cys, p.Asp444His, and p.[Ala171Thr;Asp444His]. Almost all individuals with partial biotinidase deficiency (10-30% enzyme activity) have the mutation p.Asp444His in one allele of the BTD gene in combination with a second allele.[6]
Pathophysiology
Symptoms of the deficiency are caused by the inability to reuse biotin molecules that are needed for cell growth, production of fatty acids and the metabolism of fats and amino acids. If left untreated, the symptoms can lead to later problems such as comas or death. Unless treatment is administered on a regular basis, symptoms can return at any point during the lifespan.
Diagnosis
Biotinidase deficiency can be found by genetic testing. This is often done at birth as part of newborn screening in several states throughout the United States. Results are found through testing a small amount of blood gathered through a heel prick of the infant. As not all states require that this test be done, it is often skipped in those where such testing is not required. Biotinidase deficiency can also be found by sequencing the BTD gene, particularly in those with a family history or known familial gene mutation.
Treatment
Treatment is possible but unless continued daily, problems may arise. Currently, this is done through supplementation of 5–10 mg of oral biotin a day. If symptoms have begun to show, standard treatments can take care of them, such as hearing aids for poor hearing.
Dietary Concerns
Raw eggs should be avoided in those with biotin deficiency, because egg whites contain high levels of the anti-nutrient avidin. The name avidin literally means that this protein has an "avidity" (Latin: "to eagerly long for") for biotin. Avidin binds irreversibly to biotin and this compound is then excreted in the urine.
Epidemiology
Based on the results of worldwide screening of biotinidase deficiency in 1991, the incidence of the disorder is: 5 in 137,401 for profound biotinidase deficiency
- One in 109,921 for partial biotinidase deficiency
- One in 61,067 for the combined incidence of profound and partial biotinidase deficiency
- Carrier frequency in the general population is approximately one in 120.
See also
- Biotin
- Biotin deficiency
- Multiple carboxylase deficiency
- Holocarboxylase synthetase deficiency
- 3-Methylcrotonyl-CoA carboxylase deficiency
References
- 1 2 Wolf, Barry; Norrgard, Karen; Pomponio, Robert J.; Mock, Donald M.; Secor Mcvoy, Julie R.; Fleischhauer, Kristin; Shapiro, Steven; Blitzer, Miriam G.; Hymes, Jeanne (1997). "Profound biotinidase deficiency in two asymptomatic adults". American Journal of Medical Genetics. 73 (1): 5–9. doi:10.1002/(SICI)1096-8628(19971128)73:1<5::AID-AJMG2>3.0.CO;2-U. PMID 9375914.
- 1 2 3 McVoy, Julie R. Secor; Levy, Harvey L.; Lawler, Michael; Schmidt, Michael A.; Ebers, Douglas D.; Hart, Suzanne; Pettit, Denise Dove; Blitzer, Miriam G.; Wolf, Barry (1990). "Partial biotinidase deficiency: Clinical and biochemical features". The Journal of Pediatrics. 116 (1): 78–83. doi:10.1016/S0022-3476(05)81649-X. PMID 2295967.
- ↑ Möslinger, Dorothea; Mühl, Adolf; Suormala, Terttu; Baumgartner, Regula; Stöckler-Ipsiroglu, Sylvia (2003). "Molecular characterisation and neuropsychological outcome of 21 patients with profound biotinidase deficiency detected by newborn screening and family studies". European Journal of Pediatrics. 162: S46–9. doi:10.1007/s00431-003-1351-3. PMID 14628140.
- ↑ http://www.metametrix.com/learning-center/case-studies/2004/biotin-detoxification-needs-in-cognitively-delayed-adult
- ↑ Wolf, Barry (2011). "Biotinidase Deficiency". In Pagon, Roberta A; Bird, Thomas D; Dolan, Cynthia R; Stephens, Karen; et al. GeneReviews.
- ↑ Biotinidase Deficiency (Report). Retrieved May 19, 2011.
Further reading
- Dobrowolski, Steven F.; Angeletti, Janine; Banas, Richard A.; Naylor, Edwin W. (2003). "Real time PCR assays to detect common mutations in the biotinidase gene and application of mutational analysis to newborn screening for biotinidase deficiency". Molecular Genetics and Metabolism. 78 (2): 100–7. doi:10.1016/S1096-7192(02)00231-7. PMID 12618081.
- McMahon, Robert J. (2002). "Biotin in metabolism and molecular biology". Annual Review of Nutrition. 22: 221–39. doi:10.1146/annurev.nutr.22.121101.112819. PMID 12055344.
- C. Neto, E.; Schulte, J.; Rubim, R.; Lewis, E.; Demari, J.; Castilhos, C.; Brites, A.; Giugliani, R.; et al. (2004). "Newborn screening for biotinidase deficiency in Brazil: biochemical and molecular characterizations". Brazilian Journal of Medical and Biological Research. 37 (3): 295–9. doi:10.1590/S0100-879X2004000300001. PMID 15060693.
- Weber, Peter; Scholl, Sabine; Baumgartner, E Regula (2007). "Outcome in patients with profound biotinidase deficiency: relevance of newborn screening". Developmental Medicine & Child Neurology. 46 (7): 481–4. doi:10.1111/j.1469-8749.2004.tb00509.x. PMID 15230462.
- Wolf, Barry (2003). "Biotinidase deficiency: New directions and practical concerns". Current Treatment Options in Neurology. 5 (4): 321–8. doi:10.1007/s11940-003-0038-4. PMID 12791199.
External links
![]() This article incorporates public domain material from the United States National Library of Medicine document "Genetics Home Reference".
This article incorporates public domain material from the United States National Library of Medicine document "Genetics Home Reference".