Pheochromocytoma
| Pheochromocytoma/Phaeochromocytoma | |
|---|---|
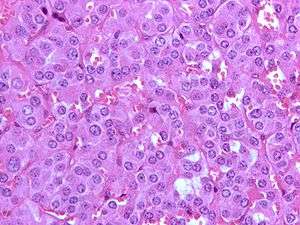 | |
| High magnification micrograph of a pheochromocytoma, showing the nested arrangement of cells (Zellballen) and stippled chromatin. H&E stain. | |
| Classification and external resources | |
| Specialty | Oncology |
| ICD-10 | D35.0, C74.1 |
| ICD-9-CM | 227.0, 194.0, 255.6 |
| ICD-O | M8700/0 |
| OMIM | 171300 |
| DiseasesDB | 9912 |
| MedlinePlus | 000340 |
| eMedicine | med/1816 radio/552 ped/1788 |
| Patient UK | Pheochromocytoma |
| MeSH | D010673 |
A pheochromocytoma (from Greek phaios "dark", chroma "color", kytos "cell", -oma "tumor") or phaeochromocytoma (PCC) is a neuroendocrine tumor of the medulla of the adrenal glands (originating in the chromaffin cells), or extra-adrenal chromaffin tissue that failed to involute after birth,[1] that secretes high amounts of catecholamines, mostly norepinephrine, plus epinephrine to a lesser extent.[2] Extra-adrenal paragangliomas (often described as extra-adrenal pheochromocytomas) are closely related, though less common, tumors that originate in the ganglia of the sympathetic nervous system and are named based upon the primary anatomical site of origin.
Signs and symptoms
The signs and symptoms of a pheochromocytoma are those of sympathetic nervous system hyperactivity,[3] including:
- Skin sensations
- Flank pain
- Elevated heart rate
- Elevated blood pressure, including paroxysmal (sporadic, episodic) high blood pressure, which sometimes can be more difficult to detect; another clue to the presence of pheochromocytoma is orthostatic hypotension (a fall in systolic blood pressure greater than 20 mmHg or a fall in diastolic blood pressure greater than 10 mmHg upon standing)
- Palpitations
- Anxiety often resembling that of a panic attack
- Diaphoresis (excessive sweating)
- Headaches – most common symptom
- Pallor
- Weight loss
- Localized amyloid deposits found microscopically
- Elevated blood glucose level (due primarily to catecholamine stimulation of lipolysis (breakdown of stored fat) leading to high levels of free fatty acids and the subsequent inhibition of glucose uptake by muscle cells. Further, stimulation of beta-adrenergic receptors leads to glycogenolysis and gluconeogenesis and thus elevation of blood glucose levels).
A pheochromocytoma can also cause resistant arterial hypertension. A pheochromocytoma can be fatal if it causes a hypertensive emergency, that is, severely high blood pressure that impairs one or more organ systems (formerly called "malignant hypertension"). This hypertension is not well controlled with standard blood pressure medications.
Not all patients experience all of the signs and symptoms listed. The most common presentation is headache, excessive sweating, and increased heart rate, with the attack subsiding in less than one hour.
Tumors may grow large, but most are smaller than 10 centimetres (4 in).
Cause
Up to 25% of pheochromocytomas may be familial. Mutations of the genes VHL, RET, NF1 (Gene 17 Neurofibromatosis type 1), SDHB and SDHD are all known to cause familial pheochromocytoma, therefore this disease may be accompanied by Von Hippel–Lindau disease, neurofibromatosis,[4] or familial paraganglioma depending on the mutation.
Pheochromocytoma is a tumor of the multiple endocrine neoplasia syndrome, type IIA and type IIB (also known as MEN IIA and MEN IIB, respectively). The other component neoplasms of that syndrome include parathyroid adenomas, and medullary thyroid cancer. Mutations in the autosomal RET proto-oncogene drives these malignancies.[5] Common mutations in the RET oncogene may also account for medullary sponge kidney as well.[6]
Pheochromocytoma linked to MEN II can be caused by RET oncogene mutations. Both syndromes are characterized by pheochromocytoma as well as thyroid cancer (thyroid medullary carcinoma). MEN IIA also presents with hyperparathyroidism, while MEN IIB also presents with mucosal neuroma.
Diagnosis
_histopathology.jpg)

The diagnosis can be established by measuring catecholamines and metanephrines in plasma (blood) or through a 24-hour urine collection. Care should be taken to rule out other causes of adrenergic (adrenalin-like) excess like hypoglycemia, stress, exercise, and drugs affecting the catecholamines like stimulants, methyldopa, dopamine agonists, or ganglion blocking antihypertensives. Various foodstuffs (e.g. coffee, tea, bananas, chocolate, cocoa, citrus fruits, and vanilla) can also affect the levels of urinary metanephrine and VMA (vanillylmandelic acid).[7]
Imaging by computed tomography or a T2 weighted MRI of the head, neck, and chest, and abdomen can help localize the tumor. Tumors can also be located using an MIBG scan, which is scintigraphy using iodine-123-marked metaiodobenzylguanidine. Even finer localization can be obtained in certain PET scan centers using PET-CT or PET-MRI with [18F] fluorodopamine[8] or FDOPA.[9]
Pheochromocytomas occur most often during young-adult to mid-adult life.
These tumors can form a pattern with other endocrine gland cancers which is labeled multiple endocrine neoplasia (MEN). Pheochromocytoma may occur in patients with MEN 2 and MEN 3 (MEN 2B). Von Hippel Lindau patients may also develop these tumors.[10]
Patients experiencing symptoms associated with pheochromocytoma should be aware that it is rare. However, it often goes undiagnosed until autopsy; therefore patients might wisely choose to take steps to provide a physician with important clues, such as recording whether blood pressure changes significantly during episodes of apparent anxiety.
Testing
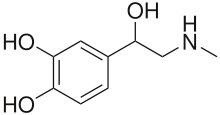

- Blood tests: Buters and others have suggested that analysis of free metanephrines (metadrenalines) (normetanephrine and metanephrine) in blood plasma is the most accurate test for detecting pheochromocytoma.
- Urine tests: Although this test is slightly less effective than plasma testing it is still considered highly effective in diagnosis. Usually the metabolites of norepinephrine and epinephrine, normetanephrine (NMN) and metanephrine (MN), are found in relatively small amounts in normal humans. The increased excretion of these metabolites is indicative of the disease, but does not completely rule out other diseases which may cause the same excretion values.
- Other Tests:
- One diagnostic test used in the past for a pheochromocytoma is to administer clonidine, a centrally-acting alpha-2 agonist used to treat high blood pressure. Clonidine mimics catecholamines in the brain, causing it to reduce the activity of the sympathetic nerves controlling the adrenal medulla. A healthy adrenal medulla will respond to the clonidine suppression test by reducing catecholamine production; the lack of a response is evidence of pheochromocytoma.
- Chromogranin A is elevated in case of pheochromocytoma.[11]
_histopathology.jpg) Micrograph of pheochromocytoma.
Micrograph of pheochromocytoma._histopathology.jpg) Micrograph of pheochromocytoma.
Micrograph of pheochromocytoma.- Micrograph of pheochromocytoma.
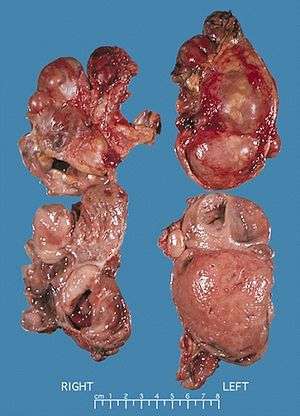 Bilateral pheochromocytoma in MEN2.
Bilateral pheochromocytoma in MEN2. Pheochromocytoma. CT abdomen.
Pheochromocytoma. CT abdomen. Pheochromocytoma. CT abdomen.
Pheochromocytoma. CT abdomen.
Tumor location
In adults, approximately 80% of pheochromocytomas are unilateral and solitary, 10% are bilateral, and 10% are extra-adrenal. In children, a quarter of tumors are bilateral, and an additional quarter are extra-adrenal. Solitary lesions inexplicably favor the right side. Although pheochromocytomas may grow to large size (>3 kg), most weigh <100 g and are <10 cm in diameter. Pheochromocytomas are highly vascular.
The tumors are made up of large, polyhedral, pleomorphic chromaffin cells. Fewer than 10% of these tumors are malignant. As with several other endocrine tumors, malignancy cannot be determined from the histologic appearance; tumors that contain large number of aneuploid or tetraploid cells, as determined by flow cytometry, are more likely to recur. Local invasion of surrounding tissues or distant metastases indicate malignancy.
Extra-adrenal pheochromocytomas: Extra-adrenal pheochromocytomas usually weigh 20 to 40 g and are <5 cm in diameter. Most are located within the abdomen in association with the celiac, superior mesenteric, and inferior mesenteric ganglia and the organ of Zuckerkandl. Approximately 10% are in the thorax, 1% are within the urinary bladder, and less than 3% are in the neck, usually in association with the sympathetic ganglia or the extracranial branches of the ninth cranial nerves.
Differential diagnosis
The differential diagnoses of pheochromocytoma include:
- Anxiety disorders, including Benzodiazepine withdrawal syndrome
- Paragangliomas
- Von Hippel–Lindau Disease
- Essential hypertension
- Hyperthyroidism
- Insulinoma
- Mercury poisoning
- Paroxysmal supraventricular tachycardia
- Renovascular hypertension
- Carcinoid[12]
Treatment
Surgical resection of the tumor is the treatment of first choice, either by open laparotomy or laparoscopy.[13] Given the complexity of perioperative management, and the potential for catastrophic intra and postoperative complications, such surgery should be performed only at centers experienced in the management of this disorder. In addition to the surgical expertise that such centers can provide, they will also have the necessary endocrine and anesthesia resources. It may also be necessary to carry out adrenalectomy, a complete surgical removal of the affected adrenal gland(s).
Either surgical option requires prior treatment with the non-specific and irreversible alpha adrenoceptor blocker phenoxybenzamine or a short acting alpha antagonist (e.g. prazosin, terazosin, or doxazosin).[14] Doing so permits the surgery to proceed while minimizing the likelihood of severe intraoperative hypertension (as might occur when the tumor is manipulated). Some authorities would recommend that a combined alpha/beta blocker such as labetalol also be given in order to slow the heart rate. Regardless, a beta-1 receptor selective beta blocker such as atenolol must never be used in the presence of a pheochromocytoma due to the risk of such a treatment leading to unopposed alpha agonism and, thus, severe and potentially refractory hypertension. However some clinical guidelines permit beta-1 blockade use together with alpha blockers during surgery for control of tachycardia.
The patient with pheochromocytoma is invariably volume depleted. In other words, the chronically elevated adrenergic state characteristic of an untreated pheochromocytoma leads to near-total inhibition of renin-angiotensin activity, resulting in excessive fluid loss in the urine and thus reduced blood volume. Hence, once the pheochromocytoma has been resected, thereby removing the major source of circulating catecholamines, a situation arises where there is both very low sympathetic activity and volume depletion. This can result in profound hypotension. Therefore, it is usually advised to "salt load" pheochromocytoma patients before their surgery. This may consist of simple interventions such as consumption of high salt food pre-operatively, direct salt replacement or through the administration of intravenous saline solution.
Prognosis
There is increased life-time risk of secondary cancers (relative risk 3.63), with a slightly increased mortality risk (1.21) according to a 2004 Swedish study. of 481 patients.[15]
Complications
The massive release of catecholamines in pheochromocytoma can cause damage to heart cells.[16] This damage may be due to either compromising the coronary microcirculation or by direct toxic effects on the heart cells.[16]
Epidemiology
Pheochromocytoma is seen in between 2–8 in 1,000,000, with approximately 1000 cases diagnosed in United States yearly. It mostly occurs in young or middle age adults, though presents earlier in hereditary cases and it is also called 10% tumor .
- About 10% of adrenal cases are bilateral (suggesting hereditary disease)
- About 10% of adrenal cases occur in children (also suggesting hereditary disease)
- About 15% are extra-adrenal (located in any orthosympathetic tissue): Of these 9% are in the abdomen, and 1% are located elsewhere. Some extra-adrenal pheochromocytomas are probably actually paragangliomas, but the distinction can only be drawn after surgical resection.
- About 11.1% of adrenal cases are malignant, but this rises to 30% for extra-adrenal cases
- About 15-20% are hereditary[17]
- About 5% are caused by VHL disease
- About 3% recur after being resected
- About 14% of affected individuals do not have arterial hypertension (Campbell's Urology)
History
In 1886, Felix Fränkel made the first description of a patient with pheochromocytoma. The term "pheochromocytoma" was first coined by Ludwig Pick, a pathologist, in 1912. In 1926, César Roux (in Switzerland) and Charles Horace Mayo (in the U.S.A.) were the first surgeons to successfully remove pheochromocytomas.
In the 1970s, Greene and Tischler derived a line of cells, called the PC12 cell line, from a rat pheochromocytoma.[18]
References
- ↑ Boulpaep, Emile L.; Boron, Walter F. (2003). Medical physiology: a cellular and molecular approach. Philadelphia: Saunders. p. 1065. ISBN 0-7216-3256-4.
- ↑ Sweeney, Ann T; Griffing, George T (August 2, 2011). "Pheochromocytoma". eMedicine.
- ↑ http://www.medicinenet.com/pheochromocytoma/page3.htm
- ↑ Goldman 2011, pp. 1194
- ↑ Online Mendelian Inheritance in Man (OMIM) MULTIPLE ENDOCRINE NEOPLASIA, TYPE IIA; MEN2A -171400
- ↑ Diouf, B.; Fary Ka, E. H.; Calender, A.; Giraud, S.; Diop, T. M. (2000). "Association of medullary sponge kidney disease and multiple endocrine neoplasia type IIA due to RET gene mutation: is there a causal relationship?". Nephrology Dialysis Transplantation. 15 (12): 2062–3. doi:10.1093/ndt/15.12.2062.
- ↑ "Catecholamines – urine – Penn State Hershey Medical Center". Pennstatehershey.adam.com. 2011-01-06. Retrieved 2013-02-23.
- ↑ "6-[18F]Fluorodopamine Positron Emission Tomographic (PET) Scanning for Diagnostic Localization of Pheochromocytoma". Hyper.ahajournals.org. Retrieved 2013-02-23.
- ↑ 6-L-18F-fluorodihydroxyphenylalanine PET in neuroendocrine tumors: basic aspects and emerging clinical applications, Journal of Nuclear Medicine 2008, 49, 573-586. DOI: 10.2967/jnumed.107.045708
- ↑ Goldman 2011, pp. 185
- ↑ Cotesta, D; Caliumi, C; Alò, P; Petramala, L; Reale, MG; Masciangelo, R; Signore, A; Cianci, R; et al. (2005). "High plasma levels of human chromogranin A and adrenomedullin in patients with pheochromocytoma". Tumori. 91 (1): 53–8. PMID 15850005.
- ↑ Giannini, A. James; Black, Henry R.; Goettsche, Roger L. (1978). Psychiatric, Psychogenic and Somatopsychic Disorders Handbook. Garden City, NY: Medical Examination. pp. 213–4. ISBN 0-87488-596-5.
- ↑ Jaroszewski, D. E.; Tessier, D. J.; Schlinkert, R. T.; Grant, C. S.; Thompson, G. B.; Van Heerden, J. A.; Farley, D. R.; Smith, S. L.; Hinder, R. A. (2003). "Laparoscopic Adrenalectomy for Pheochromocytoma". Mayo Clinic Proceedings. 78 (12): 1501–4. doi:10.4065/78.12.1501. PMID 14661679.
- ↑ Pacack, K. (2007). "Preoperative management of the pheochromocytoma patient.". J Clin Endocrinol Metab. 92 (11): 4069–79. doi:10.1210/jc.2007-1720. PMID 17989126.
- ↑ Khorram-Manesh, A.; Ahlman, H.; Nilsson, O.; Odén, A.; Jansson, S. "Mortality associated with pheochromocytoma in a large Swedish cohort". European Journal of Surgical Oncology (EJSO). 30 (5): 556–559. doi:10.1016/j.ejso.2004.03.006.
- 1 2 Goldman 2011, pp. 327
- ↑ Goldman 2011, pp. 1470
- ↑ Greene LA, Tischler AS; Tischler (1976). "Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor". Proc. Natl. Acad. Sci. U.S.A. 73 (7): 2424–8. Bibcode:1976PNAS...73.2424G. doi:10.1073/pnas.73.7.2424. PMC 430592
 . PMID 1065897.
. PMID 1065897.
Additional references
- Goldman, Lee (2011). Goldman's Cecil Medicine (24th ed.). Philadelphia: Elsevier Saunders. p. 1362. ISBN 1437727883.
External links
- MedlinePlus Overview pheochromocytoma
- GeneReviews entry on Hereditary Paraganglioma-Pheochromocytoma Syndromes
- General Information About Pheochromocytoma and Paraganglioma from the National Cancer Institute
- Neuroendocrine Tumor from the American Society of Clinical Oncology