Hydrocyanation of unsaturated carbonyl compounds
Hydrocyanation of unsaturated carbonyl compounds is a special case of the Michael reaction which can lead to β-cyanoketones, β-cyano-cyanohydrins, or vinyl cyanohydrins. Fine-tuning reaction conditions allows access to any of these products.[1]
Introduction
Hydrocyanation refers to the addition of the elements of hydrogen and cyanide across a multiple bond. When the multiple bond is polarized by an electron-withdrawing group, selective addition results. However, when the electron-withdrawing group is a carbonyl, the possibility of 1,2 (direct) addition to the carbonyl group exists. Methodological advances now permit access to both direct and conjugate addition products.
(1)
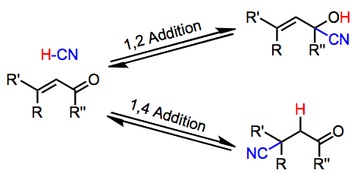
Diastereoselectivity can be achieved in prochiral, conformationally restricted substrates. Catalytic, enantioselective methods, however, are rare.[2]
Mechanism and Stereochemistry
Prevailing Mechanism
The distribution of products from conjugative hydrocyanation depends both on substrate structure and reaction conditions. Generally, however, acidic conditions favor 1,2-adducts, while basic conditions favor 1,4-adducts. As a result, under acidic conditions the cyanohydrins 2 and 4 are favored over the 1,4-adduct 3. Conditions of kinetic control also favor 1,2-adducts, while conditions of thermodynamic control favor the 1,4-adduct. Conducting the reaction at high temperatures under basic conditions thus facilitates formation of the conjugate addition product. Additions of alkali metal cyanides, for instance, lead exclusively to 1,4-addition.[3]
(2)

When organoaluminum cyanides are used as the source of the nucleophile, two distinct mechanisms are possible, depending on the organoaluminum employed. When trialkylaluminums are combined with hydrogen cyanide prior to the introduction of the carbonyl compound, a cyanoaluminate anion forms and acts as the active cyanating agent (after activation of the carbonyl compound by a proton). Formation of the dicyano compound is rapid under these conditions, but conversion back to the keto form occurs under basic workup.[4]
(3)
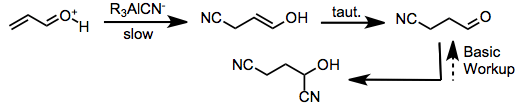
When neutral, pre-formed dialkylaluminum cyanides are used, both carbonyl activation and nucleophilic attack of cyanide are facilitated by the organoaluminum reagent. 1,2-Addition is rapid in this case but is also reversible, and slow but irreversible 1,4-addition eventually leads to the conjugate addition product.[5]
Control of 1,2- versus 1,4-Addition
Direct versus conjugate addition depends on a number of factors. The structure of the carbonyl compound is important, as β-substitution and nearby angular substituents sterically disfavor 1,4-addition. The nature of the cyanating reagent also plays a role—when dialkylaluminum cyanides are used, the development of a six-membered transition state with s-cis enones may favor 1,4-addition. "Bare" cyanating reagents such as alkali metal cyanides and cyanoaluminates tend to add in a 1,4 fashion, whereas Lewis acidic cyanides, such as TMSCN, favor 1,2-addition.
Scope and Limitations
Hydrogen cyanide alone is not reactive enough to add to carbonyl groups; as a result, base catalysis is necessary. Conjugative hydrocyanation is limited by some side reactions and the strongly basic conditions typically employed. Product hydrolysis should be expected in reactions of alkali metal cyanides. Epimerization at the α-position of carbonyls, double bond isomerization, and α-acetoxy rearrangements have all been observed as side reactions under basic conditions. Nucleophilic solvents may add competitively to the carbonyl substrate. Michael addition of cyanated products to the unsaturated carbonyl starting material is also an important side reaction.[6]
Acetone cyanohydrin can be used to reversibly generate the cyanide anion.[7]
(4)
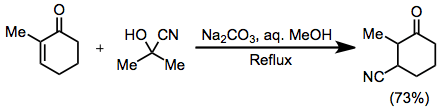
Beyond a requirement for base-stable functionality, a wide variety of carbonyl substrates undergo conjugative hydrocyanation. Acetylenic substrates undergo the reaction; however the scope of this reaction is limited and yields are often low.[8]
(5)
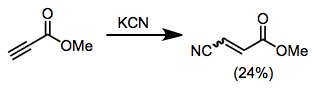
1,4-Addition to imines has been observed in a few cases, although imines are often base labile.[9]
(6)

Esters,[10] nitriles[11] and other carbonyl derivatives also undergo conjugative hydrocyanation.
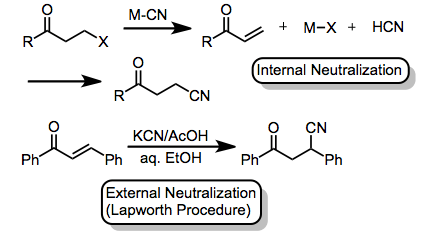
When alkali metal cyanides are used, at least partial neutralization of the reaction medium is usually necessary to provide desired products. Neutralization can be accomplished through an acidic group on the substate itself (internal neutralization).[12] or through the addition of an external acid (external neutralization). Acetic acid is commonly used for this purpose, in a procedure pioneered by Lapworth.[13]
(7)
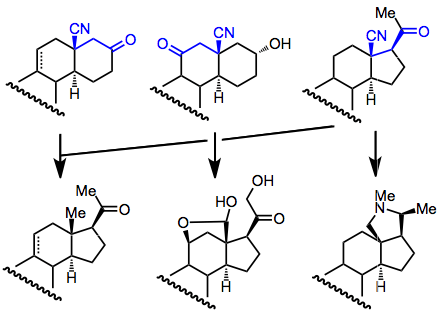
Synthetic Applications
Conjugative hydrocyanation has been heavily used for steroid synthesis. Originally, it was used to prepare the steroidal D ring, but other approaches have also been employed.[14] Diastereoselectivity is generally high in these addition reactions, and the resulting β-cyano carbonyl compounds can be converted to a number of steroidal products.
(8)
Comparison with Other Methods
Conjugate hydrocyanation is most comparable to the conjugate addition of alkyl groups mediated by organocopper species. Whereas copper conjugate addition is best suited for the introduction of alkyl groups at the β-position, hydrocyanation works well for the introduction of functional groups that may undergo further transformations. Cyanating reagents are much less sensitive to the steric environment of the β-position than organocopper reagents,[15] and organoaluminum cyanating reagents are not sensitive to halogens while organocopper reagents are.
Experimental Conditions and Procedure
Typical Conditions
Cyanation reagents are highly toxic and should be handled with care. Most cyanating agents are commercially available, but a number of procedures are available for their preparation from alkali metal cyanides. Concentration, solvent, temperature, and time should all be optimized to minimize by-products. For alkali metal cyanide methods and methods employing acetone cyanohydrin, alcoholic solvents are commonly used. Special workup procedures are needed for reactions of imines, acid chlorides, and acid cyanides. Generally however, methods employing organoaluminums should involve a very careful basic workup (to avoid hydrolysis or epimerization of products). Chromatography should be carried out under acidic conditions to avoid nitrile hydrolysis (10% acetic acid is sometimes added).
Example Procedure[16]
(9)
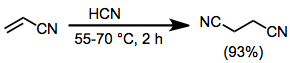
To 300 g (5.65 mol) of acrylonitrile containing 3 g (0.054 mol) of potassium cyanide was added at once 50 g (1.85 mol) of liquid hydrogen cyanide. When reaction did not start, the mixture was warmed to 30° for a short period. Soon an exothermic reaction began, and the reaction temperature was maintained at 55–60° with ice cooling. After the exothermic reaction subsided, 103 g (3.8 mol) of liquid hydrogen cyanide was added dropwise with cooling. After it had been heated at 60–70° for 2 hours, the resulting dark-brown reaction mixture was distilled under reduced pressure to give 420 g (93%) of succinonitrile as a colorless oil, bp 136° (10 mm), which solidified (mp 55°) on cooling. 1H NMR (CDCl3): δ 2.76 (s, 4H).[17]
References
- ↑ Nagata, W.; Yoshioka, M. Org. React. 1977, 25, 255. doi:10.1002/0471264180.or025.03
- ↑ Sammis, G.M.; Danjo, H.; Jacobsen, E.N. J. Am. Chem. Soc. 2004, 126, 9928.
- ↑ Mowry, T. Chem. Rev. 1948, 42, 189.
- ↑ Bonitz, E. Chem. Ber. 1955, 88, 742.
- ↑ Nagata, W. Proc. R. A. Welch Foundation on Chem. Res. 1973, 17, 185.
- ↑ Birch, J. ; Corrie, T. ; Macdonald, L. ; Rao, S. J. Chem. Soc., Perkin I, 1972, 1186.
- ↑ Nazarov, N. ; Zav'yalov, I. J. Gen. Chem. USSR (Engl. Transl.) 1954, 24, 475 [C.A., 49, 6139f (1955)].
- ↑ Kurtz, P. Ann. Chem. 1951, 572, 23.
- ↑ Nagata, W. ; Yoshioka, M. ; Okumura, T. ; Murakami, M. J. Chem. Soc., C, 1970, 2355.
- ↑ Allen, H. ; Johnson, B. Org. Synth. 1963, Coll. Vol. IV, 804.
- ↑ Kurtz, P. Ann. Chem. 1951, 572, 23.
- ↑ Crabbé, P.; Pérez, M.; Vera, G. Can. J. Chem. 1963, 41, 156.
- ↑ Lapworth, A. ; Wechsler, E. J. Chem. Soc. 1910, 97, 38.
- ↑ Nagata, W. ; Terasawa, T. ; Hirai, S. ; Takeda, K. Tetrahedron Lett., 1960, 17, 27.
- ↑ Posner, H. Org. React. 1972, 19, 1.
- ↑ Kurtz, P. Ann. Chem. 1951, 572, 23.
- ↑ Roh, J. Synthesis 2009, 2175.