Human mitochondrial molecular clock
The human mitochondrial molecular clock is the rate at which mutations have been accumulating in the mitochondrial genome of hominids during the course of human evolution. The archeological record of human activity from early periods in human prehistory is relatively limited and its interpretation has been controversial. Because of the uncertainties from the archeological record, scientists have turned to molecular dating techniques in order to refine the timeline of human evolution. A major goal of scientists in the field is to develop an accurate hominid mitochondrial molecular clock which could then be used to confidently date events that occurred during the course of human evolution.
Estimates of the mutation rate of human mitochondrial DNA (mtDNA) vary greatly depending on the available data and the method used for estimation. The two main methods of estimation, phylogeny based methods and pedigree based methods, have produced mutation rates that differ by almost an order of magnitude. Current research has been focused on resolving the high variability obtained from different rate estimates.
Rate variability
A major assumption of the molecular clock theory is that mutations within a particular genetic system occur at a statistically uniform rate and this uniform rate can be used for dating genetic events. In practice the assumption of a single uniform rate is an oversimplification. Though a single mutation rate is often applied, it is often a composite or an average of several different mutation rates.[1] Many factors influence observed mutation rates and these factors include the type of samples, the region of the genome studied and the time period covered.
Actual vs. observed rates
The rate at which mutations occur during reproduction, the germline mutation rate, is thought to be higher than all observed mutation rates, because not all mutations are successfully passed down to subsequent generations.[2] MtDNA is only passed down along the matrilineal line, and therefore mutations passed down to sons are lost. Random genetic drift may also cause the loss of mutations. For these reasons, the actual mutation rate will not be equivalent to the mutation rate observed from a population sample.[2]
Population size
Population dynamics are believed to influence observed mutation rates. When a population is expanding, more germline mutations are preserved in the population. As a result, observed mutation rates tend to increase in an expanding population. When populations contract, as in a population bottleneck, more germline mutations are lost. Population bottlenecks thus tend to slow down observed mutation rates. Since the emergence of the species homo sapiens about 200,000 years ago, human population have expanded from a few thousand individuals living in Africa to over 6.5 billion all over the world. However the expansion has not been uniform, the history of human populations may have consisted of both bottlenecks and expansions.[3]
Structural variability
The mutation rate across the mitochondrial genome is not uniformly distributed. Certain regions of the genome are known to mutate more rapidly than others. The Hypervariable regions are known to be highly polymorphic relative to other parts of the genome.
The rate at which mutations accumulate in coding and non-coding regions of the genome also differs as mutations in the coding region are subject to purifying selection. For this reason, some studies avoid coding region or synonymous mutations when calibrating the molecular clock. Loogvali et al. (2009) only consider synonymous mutations, they have recalibrated the molecular clock of human mtDNA as 7990 years per synonymous mutation over the mitochondrial genome.[1] Soares et al. (2009) consider both coding and non-coding region mutations to arrive at a single mutation rate, but apply a correction factor to account for selection in the coding region.
Temporal variability
The mutation rate has been observed to vary with time. Mutation rates within the human species are faster than those observed along the human-ape lineage. The mutation rate is also thought to be faster in recent times, since the beginning of the Holocene 11,000 years ago.[1][3][4]
Parallel mutations and saturation
Parallel mutation (sometimes referred to as Homoplasy) or convergent evolution occurs when separate lineages have the same mutation independently occur at the same site in the genome. Saturation occurs when a single site experiences multiple mutations. Parallel mutations and saturation result in the underestimation of the mutation rate because they are likely to be overlooked.[2]
Heteroplasmy
Individuals affected by heteroplasmy have a mixture of mtDNA types, some with new mutations and some without. The new mutations may or may not be passed down to subsequent generations. Thus the presence of heteroplasmic individuals in a sample may complicate the calculation of mutation rates.[2][5]
Methods
Pedigree based
Pedigree methods estimate the mutation rate by comparing the mtDNA sequences of a sample of parent/offspring pairs or analyzing mtDNA sequences of individuals from a deep-rooted genealogy. The number of new mutations in the sample is counted and divided by the total number of parent-to-child DNA transmission events to arrive at a mutation rate.[3][5]
Phylogeny based
Phylogeny based methods are estimated by first reconstructing the haplotype of the most recent common ancestor (MRCA) of a sample of two or more genetic lineages. A requirement is that the time to the most recent common ancestor (TMRCA) of the sample of lineages must already be known from other independent sources, usually the archeological record. The average number of mutations that have accumulated since the MRCA is then computed and divided by the TMRCA to arrive at the mutation rate. The human mutation rate is usually estimated by comparing the sequences of modern humans and chimpanzees and then reconstructing the ancestral haplotype of the chimpanzee-human common ancestor. According to the paleontological record the last common ancestor of humans may have lived around 6 million years ago.[3]
Pedigree vs. Phylogeny comparison
Rates obtained by pedigree methods are about 10 times faster than those obtained by phylogenetic methods. Several factors acting together may be responsible for this difference. As pedigree methods record mutations in living subjects, the mutation rates from pedigree studies are closer to the germline mutation rate. Pedigree studies use genealogies that are only a few generations deep whereas phylogeny based methods use timescales that are thousands or millions of years deep. According to Henn et al. 2009, phylogeny based methods take into account events that occur over long time scales and are thus less affected by stochastic fluctuations. Howell et al. 2003 suggests that selection, saturation, parallel mutations and genetic drift are responsible for the differences observed between pedigree based methods and phylogeny based methods.
Estimating based on AMH archaeology
| Study | Sequence type | TAnchor (location) | Referencing method (correction method) |
| Cann, Stoneking & Wilson (1987) | Restriction fragments | 40, 30, and 12 Ka (Australia, New Guinea New World) | archaeologically defined migrations matched with estimated sequence divergence rates |
| Endicott & Ho (2008) | Genomic | 40 to 55 Ka (Papua New Guinea) 14.5 to 21.5 Ka (Haps H1 and H3) | PNG following Haplogroup P |
Anatomical modern humans (AMH) spread out of Africa and over a large area of Eurasia and left artifacts along the northern coast of the Southwest, South, Southeast and East Asia. Cann, Stoneking & Wilson (1987) did not rely on a predicted TCHLCA to estimate single-nucleotide polymorphism (SNP) rates. Instead, they used evidence of colonization in Southeast Asia and Oceania to estimate mutation rates. In addition they used RFLP technology (Restriction fragment length polymorphism) to examine differences between DNA. Using these techniques this group came up with a TMRCA of 140,000 to 290,000 years. It should be noted however that Cann et al. (1987) estimated the TMRCA of humans to be approximately 210 ky and the most recent estimates Soares et al. 2009 (using 7 million year chimpanzee human mtDNA MRCA) differ by only 9%, which is relatively close considering the wide confidence range for both estimates and calls for more ancient TCHLCA.
Endicott & Ho (2008) have reevaluated the predicted migrations globally and compared those to the actual evidence. This group used the coding regions of sequences. They postulate that the molecular clock based on chimp-human comparisons is not reliable, particularly in predicting recent migrations, such as founding migrations into Europe, Australia, and the Americans. With this technique this group came up with a TMRCA of 82,000 to 134,000 years.
Estimating based on CHLCA
Because chimps and humans share a matrilineal ancestor, establishing the geological age of that last ancestor allows the estimation of the mutation rate. The chimp-human last common ancestor (CHLCA) is frequently applied as an anchor for mt-TMRCA studies with ranges between 4 and 13 million years cited in the literature.[6] This is one source of variation in the time estimates. The other weakness is the non-clocklike accumulation of SNPs, would tend to make more recent branches look older than they actually are.[7]
| Regions(s) | Subregions (or site within codon) | SNP rate (per site * year) | |
| Control region | HVR I | 1.6 × 10−7 | |
| HVR II | 2.3 × 10−7 | ||
| remaining | 1.5 × 10−8 | ||
| Protein- coding | (1st and 2nd) | 8.8 × 10−9 | |
| (3rd) | 1.9 × 10−8 | ||
| DNA encoding rRNA (rDNA) | 8.2 × 10−9 | ||
| DNA encoding tRNA (tDNA) | 6.9 × 10−9 | ||
| other | 2.4 × 10−8 | ||
| TCHLCA assumed 6.5 Ma, relative rate to 1st & 2nd codons | |||
These two sources may balance each other or amplify each other depending on the direction of the TCHLCA error. There are two major reasons why this method is widely employed. First the pedigree based rates are inappropriate for estimates for very long periods of time. Second, while the archaeology anchored rates represent the intermediate range, archaeological evidence for human colonization often occurs well after colonization. For example, colonization of Eurasia from west to east is believed to have occurred along the Indian Ocean. However, the oldest archaeological sites that also demonstrate anatomically modern humans (AMH) are in China and Australia, greater than 42,000 years in age. However the oldest Indian site with AMH remains is from 34,000 years, and another site with AMH compatible archaeology is in excess of 76,000 years in age.[7] Therefore, application of the anchor is a subjective interpretation of when humans were first present.
A simple measure the sequence divergence between humans and chimps by observing the SNPs. Given that the mitogenome is about 16553 base pairs in length (each base-pair which can be aligned with known references is called a site).[8] The formula is:

The '2' in the denominator is derived from the 2 lineages, human and chimpanzee, that split from the CHLCA. Ideally it represents the accumulation of mutations on both lineages but in different positions (SNPs). As long as the number of SNP observed approximates the number of mutations this formula works well. However, at rapidly evolving sites mutations are obscured by saturation affects. Sorting positions within the mitogenome by rate and compensating for saturation are alternative approaches.[9]
Because the TCHLCA is subject to change with more paleontological information, the equation described above allows the comparison of TMRCA from different studies.
| Study | Sequence type | TCHLCA (sorting time) | Referencing method (correction method) |
| Vigilant et al. (1991) | HVR | 4 to 6 Ma | CH transversions, (15:1 transition:transversion) |
| Ingman et al. (2000) | genomic (not HVR) | 5 Ma | CH genomic comparison |
| Endicott & Ho (2008) | genomic (not HVR) | 5 to 7.5 Ma | CH (relaxed rate, rate-class defined) |
| Gonder et al. (2007) | genomic (not HVR) | 6.0 Ma (+ 0.5 Ma) | CH (rate class defined) |
| Mishmar et al. (2003) | genomic (not HVR) | 6.5 Ma (+ 0.5 Ma) | CH (rate class defined) |
| Soares et al. (2009) | genomic | 6.5Ma (+ 0.5 Ma) | CHLCA anchored, (Examined selection by Ka/(Ks + k)) |
| Chimpanzee to Human = CH, LCA = last common ancestor | |||
Early, HVR, sequence-based methods
To overcome the effects of saturation, HVR analysis relied on the transversional distance between humans and chimpanzees.[10] A transition to transversion ratio was applied to this distance to estimate sequence divergence in the HVR between chimpanzees and humans, and divided by an assumed TCHLCA of 4 to 6 million years.[11] Based on 26.4 substitutions between chimpanzee and human and 15:1 ratio, the estimated 396 transitions over 610 base-pairs demonstrated sequence divergence of 69.2% (rate * TCHLCA of 0.369), producing divergence rates of roughly 11.5% to 17.3% per million years.
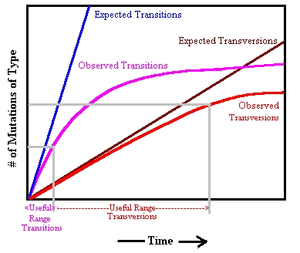
Vigilant et al. (1991) also estimated the sequence divergence rate for the sites in the rapidly evolving HVR I and HVR II regions. As noted in the table above, the rate of evolution is so high that site saturation occurs in direct chimpanzee and human comparisons. Consequently, this study used transversions, which evolve at a slower rate than the more common transition polymorphisms. Comparing chimp and human mitogenomes, they noted 26.4 transversions within the HVR regions, however they made no correction for saturation. As more HVR sequence was obtained following this study, it was noted that the dinucleotide site CRS:16181-16182 experienced numerous transversions in parsimony analysis, many of these were considered to be sequencing errors. However the sequencing of Feldhofer I Neanderthal revealed that there was also a transversion between humans and Neanderthals at this site.[12] In addition, Soares et al. (2009) noted three sites in which recurrent transversions had occurred in human lineages, two of which are in HVR I, 16265 (12 occurrences) and 16318(8 occurrences).[note 1] Therefore, 26.4 transversions was an underestimate of the likely number of transversion events. The year 1991 study also used a transition-to-transversion ratio from the study of old world monkeys of 15:1. However, examination of chimp and gorilla HVR reveals a rate that is lower, and the examination of humans places the rate at 34:1.[6] Therefore, this study underestimated that level of sequence divergence between chimpanzee and human. The estimated sequence divergence 0.738/site (includes transversions) is significantly lower than the ~2.5 per site suggested by Soares et al. (2009). These two errors would result in an overestimate of the human mitochondrial TMRCA. However, they failed to detect the basal L0 lineage in the analysis and also failed to detect recurrent transitions in many lineages, which also underestimate the TMRCA. Also, Vigilant et al. (1991) used a more recent CHLCA anchor of 4 to 6 million years.
Coding region sequence based methods
| African mtDNA haplogroups | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Partial coding region sequence originally supplemented HVR studies because complete coding region sequence was uncommon. There were suspicions that the HVR studies had missed major branches based on some earlier RFLP and coding region studies. Ingman et al. (2000) was the first study to compare genomic sequences for coalescence analysis. Coding region sequence discriminated M and N haplogroups and L0 and L1 macrohaplogroups. Because the genomic DNA sequencing resolved the two deepest branches it improved some aspects estimating TMRCA over HVR sequence alone. Excluding the D-loop and using a 5-million-year TCHLCA, Ingman et al. (2000) estimated the mutation rate to be 1.70 × 10−8 per site per year (rate * TCHLCA = 0.085, 15,435 sites).
However, coding region DNA has come under question because coding sequences are either under purifying selection to maintain structure and function, or under regional selection to evolve new capacities.[13] The problem with mutations in the coding region has been described as such: mutations occurring in the coding region that are not lethal to the mitochondria can persist but are negatively selective to the host; over a few generations these will persist, but over thousands of generations these slowly are pruned from the population, leaving SNPs.[6] However, over thousands of generations regionally selective mutations may not be discriminated from these transient coding region mutations. The problem with rare mutations in the human mitogenomes is significant enough to prompt a half-dozen recent studies on the matter.
Ingman et al. (2000) estimated the non-D loop region evolution 1.7 × 10−8 per year per site based on 53 non-identical genomic sequence overrepresenting Africa in a global sample. Despite this over-representation, the resolution of the L0 subbranches was lacking and one other deep L1 branches has been found. Despite these limitations that sampling was adequate for the hallmark study. Today, L0 is restricted to African populations, whereas L1 is the ancestral haplogroup of all non-Africans, as well as most Africans. Mitochondrial Eve's sequence can be approximated by comparing a sequence from L0 with a sequence from L1. By reconciling the mutations in L0 and L1. The mtDNA sequences of contemporary human populations will generally differ from Mitochondrial Eve's sequence by about 50 mutations.[14][15] Mutation rates were not classified according to site (other than excluding the HVR reigons). The TCHLCA used in the year 2000 study of 5 Ma was also lower than values used in the most recent studies.
Estimates from ancient DNA
Since it has become possible to sequence large numbers of ancient mitogenomes, several studies have estimated the mitochondrial mutation rate by measuring how many more mutations on average have accumulated in modern (or later) genomes compared to ancient (or earlier) ones descending from the same phylogenetic node. These studies have obtained similar results: central estimates for the whole chromosome, in substitutions per site per year: 2.47 × 10−8;[16] 2.14 × 10−8;[17] 2.53 × 10−8;[18] and 2.74 × 10−8.[19]
Inter-comparing rates and studies
Molecular clocking of mitochondrial DNA has been criticized because of its inconsistent molecular clock.[20][21][22] A retrospective analysis of any pioneering process will reveal inadequacies. With mitochondrial the inadequacies are the argument from ignorance of rate variation and overconfidence concerning the TCHLCA of 5 Ma. Lack of historical perspective might explain the second issue, the problem of rate variation is something that could only be resolved by the massive study of mitochondria that followed. The number of HVR sequences that have accumulated from 1987 to 2000 increased by magnitudes. Soares et al. (2009) used 2196 mitogenomic sequences and uncovered 10,683 substitution events within these sequences. Eleven of 16560 sites in the mitogenome produced greater than 11% of all the substitutions with statistically significant rate variation within the 11 sites.[note 2] They argue that there is a neutral-site mutation rate which is a magnitude slower than rate observed for the fastest site, CRS 16519. Consequently, purifying selection aside, the rate of mutation itself varies between sites, with a few sites much more likely to undergo new mutations relative to others.[23] Soares et al. (2009) noted two spans of DNA, CRS 2651-2700 and 3028-3082, that had no SNPs within the 2196 mitogenomic sequences.
|
Phylogenetic tree of human mitochondrial DNA (mtDNA) haplogroups | |||||||||||||||||||||||||||||||||||||
| Mitochondrial Eve (L) | |||||||||||||||||||||||||||||||||||||
| L0 | L1–6 | ||||||||||||||||||||||||||||||||||||
| L1 | L2 | L3 | L4 | L5 | L6 | ||||||||||||||||||||||||||||||||
| M | N | ||||||||||||||||||||||||||||||||||||
| CZ | D | E | G | Q | O | A | S | R | I | W | X | Y | |||||||||||||||||||||||||
| C | Z | B | F | R0 | pre-JT | P | U | ||||||||||||||||||||||||||||||
| HV | JT | K | |||||||||||||||||||||||||||||||||||
| H | V | J | T | ||||||||||||||||||||||||||||||||||
Notes
Footnotes
- 1 2 3 Loogvali et al. (2009)
- 1 2 3 4 Howell, N; Smejkal, CB; MacKey, DA; Chinnery, PF; Turnbull, DM; Herrnstadt, C (2003), "The Pedigree Rate of Sequence Divergence in the Human Mitochondrial Genome: There Is a Difference Between Phylogenetic and Pedigree Rates", American Journal of Human Genetics, 72 (3): 659–70, doi:10.1086/368264, PMC 1180241
 , PMID 12571803.
, PMID 12571803. - 1 2 3 4 Henn et al. (2009)
- ↑ Ho SY, Phillips MJ, Cooper A, Drummond AJ (2005), "Time Dependency of Molecular Rate Estimates and Systematic Overestimation of Recent Divergence Times", Molecular Biology and Evolution, 22 (7): 1561–8, doi:10.1093/molbev/msi145, PMID 15814826.
- 1 2 Sigurðardóttir et al. (2000)
- 1 2 3 Soares et al. (2009)
- 1 2 see: Endicott et al. (2009)
- ↑ Ingman et al. (2000)
- ↑ See: Gonder et al. (2007),Soares et al. (2009)
- ↑ Vigilant et al. (1989)
- ↑ Vigilant et al. (1991)
- ↑ Krings et al. (1997)
- ↑ see: Suissa et al. (2009), Balloux et al. (2009)
- ↑ Gonder et al. (2007)
- ↑ Behar DM; Villems R; Soodyall H; Blue-Smith J; Pereira L; Metspalu E; Scozzari R; Makkan H; Tzur S; Comas D, D; Bertranpetit J; Quintana-Murci L; Tyler-Smith C; Wells RS; Rosset S; Genographic Consortium (May 2008). "The dawn of human matrilineal diversity". American Journal of Human Genetics. pp. 1130–40. doi:10.1016/j.ajhg.2008.04.002. PMC 2427203. PMID 18439549.
- ↑ Fu Q, Mittnick A, et al. (April 2013), "A revised timescale for human evolution based on ancient mitochondrial genomes", Curr. Biol., 23 (7): 553–559, doi:10.1016/j.cub.2013.02.044.
- ↑ Rieux A, Eriksson A, et al. (August 2014), "Improved calibration of the human mitochondrial clock using ancient genomes", Mol. Biol. Evol., 31 (10): 2780–2792, doi:10.1093/molbev/msu222.
- ↑ Fu Q, Li H, et al. (October 2014), "Genome sequence of a 45,000-year-old modern human from western Siberia", Nature, 514 (7523): 445–449, doi:10.1038/nature13810.
- ↑ Posth C, Renaud G, et al. (March 2016), "Pleistocene mitochondrial genomes suggest a single major dispersal of non-Africans and a Late Glacial population turnover in Europe", Curr. Biol., 26 (6): 827–833, doi:10.1016/j.cub.2016.01.037.
- ↑ Ho SY, Larson G (February 2006), "Molecular clocks: when times are a-changin'", Trends Genet., 22 (2): 79–83, doi:10.1016/j.tig.2005.11.006, PMID 16356585.
- ↑ Gibbons A (January 1998), "Calibrating the mitochondrial clock", Science, 279 (5347): 28–9, doi:10.1126/science.279.5347.28, PMID 9441404.
- ↑ Santos C, Sierra B, Alvarez L, Ramos A, Fernández E, Nogués R, Aluja MP (2008), "Frequency and pattern of heteroplasmy in the control region of human mitochondrial DNA", J Mol Evol., 67 (2): 191–200, doi:10.1007/s00239-008-9138-9, PMID 18618067.
- ↑ Excoffier L, Yang Z (October 1999), "Substitution rate variation among sites in mitochondrial hypervariable region I of humans and chimpanzees", Mol. Biol. Evol, 16 (10): 1357–68, doi:10.1093/oxfordjournals.molbev.a026046, PMID 10563016.
References
- Atkinson, QD; Gray, RD; Drummond, AJ (January 2009), "Bayesian coalescent inference of major human mitochondrial DNA haplogroup expansions in Africa", Proceedings of the Royal Society B: Biological Sciences, 276 (1655): 367–73, doi:10.1098/rspb.2008.0785, PMC 2674340, PMID 18826938
- Balloux, F; Handley, LJ; Jombart, T; Liu, H; Manica, A (2009), "Climate shaped the worldwide distribution of human mitochondrial DNA sequence variation", Proc Biol Sci., 276 (1672): 3447–55, doi:10.1098/rspb.2009.0752, PMC 2817182, PMID 19586946
- Behar DM; Villems R; Soodyall H; Blue-Smith J; Pereira L; Metspalu E; Scozzari R; Makkan H; Tzur S; Comas D, D; Bertranpetit J; Quintana-Murci L; Tyler-Smith C; Wells RS; Rosset S; Genographic Consortium (May 2008). "The dawn of human matrilineal diversity". American Journal of Human Genetics. pp. 1130–40. doi:10.1016/j.ajhg.2008.04.002. PMC 2427203. PMID 18439549.
- Cann, RL; Stoneking, M; Wilson, AC (1987), "Mitochondrial DNA and human evolution", Nature, 325 (6099): 31–6, doi:10.1038/325031a0, PMID 3025745
- Cox, MP (August 2008), "Accuracy of molecular dating with the rho statistic: deviations from coalescent expectations under a range of demographic models", Hum. Biol., 80 (4): 335–57, doi:10.3378/1534-6617-80.4.335, PMID 19317593
- Endicott, P; Ho, SY (April 2008), "A Bayesian evaluation of human mitochondrial substitution rates", American Journal of Human Genetics, 82 (4): 895–902, doi:10.1016/j.ajhg.2008.01.019, PMC 2427281, PMID 18371929
- Endicott, P; Ho, SY; Metspalu, M; Stringer, C (September 2009), "Evaluating the mitochondrial timescale of human evolution", Trends Ecol. Evol. (Amst.), 24 (9): 515–21, doi:10.1016/j.tree.2009.04.006, PMID 19682765
- Excoffier, L; Yang, Z (October 1999), "Substitution rate variation among sites in mitochondrial hypervariable region I of humans and chimpanzees", Mol. Biol. Evol., 16 (10): 1357–68, doi:10.1093/oxfordjournals.molbev.a026046, PMID 10563016
- Felsenstein, J (April 1992), "Estimating effective population size from samples of sequences: inefficiency of pairwise and segregating sites as compared to phylogenetic estimates", Genet. Res., 59 (2): 139–47, doi:10.1017/S0016672300030354, PMID 1628818
- Gonder, MK; Mortensen, HM; Reed, FA; de Sousa, A; Tishkoff, SA (December 2007), "Whole-mtDNA genome sequence analysis of ancient African lineages", Mol. Biol. Evol, 24 (3): 757–68, doi:10.1093/molbev/msl209, PMID 17194802
- Henn, B. M.; Gignoux, C. R.; Feldman, M. W.; Mountain, J. L. (2009), "Characterizing the Time Dependency of Human Mitochondrial DNA Mutation Rate Estimates", Molecular Biology and Evolution, 26 (1): 217–230, doi:10.1093/molbev/msn244, PMID 18984905
- Ingman, M; Kaessmann, H; Pääbo, S; Gyllensten, U (December 2000), "Mitochondrial genome variation and the origin of modern humans", Nature, 408 (6813): 708–13, doi:10.1038/35047064, PMID 11130070
- Kaessmann, H; Pääbo, S (January 2002), "The genetical history of humans and the great apes", J. Intern. Med., 251 (1): 1–18, doi:10.1046/j.1365-2796.2002.00907.x, PMID 11851860
- Kimura, M (1962), "On the Probability of Fixation of Mutant Genes in a Population", Genetics, 47: 713–719, PMC 1210364, PMID 14456043
- Loogvali, Eva-Liis; Kivisild, Toomas; Margus, Tõnu; Villems, Richard (2009), O'Rourke, Dennis, ed., "Explaining the Imperfection of the Molecular Clock of Hominid Mitochondria", PLoS ONE, 4 (12): e8260, doi:10.1371/journal.pone.0008260, PMC 2794369, PMID 20041137
- Scherer, S; Loewe, L (November 1997), "Mitochondrial Eve: The Plot Thickens", Trends in Ecology & Evolution, 12 (11): 422–3, doi:10.1016/S0169-5347(97)01204-4
- Maca-Meyer, N; González, AM; Larruga, JM; Flores, C; Cabrera, VM (2001), "Major genomic mitochondrial lineages delineate early human expansions", BMC Genet., 2: 13, doi:10.1186/1471-2156-2-13, PMC 55343, PMID 11553319
- Nielsen, R; Beaumont, MA (March 2009), "Statistical inferences in phylogeography", Mol. Ecol., 18 (6): 1034–47, doi:10.1111/j.1365-294X.2008.04059.x, PMID 19207258
- Oppenheimer, Stephen (2004), The Real Eve: Modern Man's Journey Out of Africa, New York, NY: Carroll & Graf, ISBN 0-7867-1334-8
- Parsons, TJ; Muniec, DS; Sullivan, K; Woodyatt, N; Alliston-Greiner, R; Wilson, MR; Berry, DL; Holland, KA; Weedn, VW; Holland, MM (April 1997), "A high observed substitution rate in the human mitochondrial DNA control region", Nat. Genet., 15 (4): 363–8, doi:10.1038/ng0497-363, PMID 9090380
- Sigurðardóttir, S; Helgason, A; Gulcher, JR; Stefansson, K; Donnelly, P (2000), "The Mutation Rate in the Human mtDNA Control Region", American Journal of Human Genetics, 66 (5): 1599–609, doi:10.1086/302902, PMC 1378010, PMID 10756141
- Soares, P; Ermini, L; Thomson, N; Mormina, M; Rito, T; Röhl, A; Salas, A; Oppenheimer, S; Macaulay, V; Richards, MB (June 2009), "Correcting for purifying selection: an improved human mitochondrial molecular clock", American Journal of Human Genetics, 84 (6): 740–59, doi:10.1016/j.ajhg.2009.05.001, PMC 2694979, PMID 19500773
- Suissa, S; Wang, Z; Poole, J; Wittkopp, S; Feder, J; Shutt, TE; Wallace, DC; Shadel, GS; Mishmar, D (2009), Desalle, R, ed., "Ancient mtDNA genetic variants modulate mtDNA transcription and replication", PLoS Genet., 5 (5): e1000474, doi:10.1371/journal.pgen.1000474, PMC 2673036, PMID 19424428
- Takahata, N (January 1993), "Allelic genealogy and human evolution", Mol. Biol. Evol., 10 (1): 2–22, PMID 8450756
- Vigilant, L; Pennington, R; Harpending, H; Kocher, TD; Wilson, AC (December 1989), "Mitochondrial DNA sequences in single hairs from a southern African population", Proc. Natl. Acad. Sci. U.S.A., 86 (23): 9350–4, doi:10.1073/pnas.86.23.9350, PMC 298493, PMID 2594772
- Vigilant, L; Stoneking, M; Harpending, H; Hawkes, K; Wilson, AC (September 1991), "African populations and the evolution of human mitochondrial DNA", Science, 253 (5027): 1503–7, doi:10.1126/science.1840702, PMID 1840702
- Watson E, Forster P, Richards M, Bandelt HJ (September 1997), "Mitochondrial footprints of human expansions in Africa", American Journal of Human Genetics, 61 (3): 691–704, doi:10.1086/515503, PMC 1715955, PMID 9326335