Glycogen storage disease type I
| Glycogen storage disease type I | |
|---|---|
| Classification and external resources | |
| Specialty | endocrinology |
| ICD-10 | E74.0 |
| ICD-9-CM | 271.0 |
| OMIM | 232200 232240 |
| DiseasesDB | 5284 |
| MedlinePlus | 000338 |
| eMedicine | ped/2416 |
| GeneReviews | |
Glycogen storage disease type I (GSD I) or von Gierke disease, is the most common of the glycogen storage diseases. This genetic disease results from deficiency of the enzyme glucose-6-phosphatase, and has an incidence in the American population of approximately 1 in 50,000[1] to 100,000[2] births.
The deficiency impairs the ability of the liver to produce free glucose from glycogen and from gluconeogenesis. Since these are the two principal metabolic mechanisms by which the liver supplies glucose to the rest of the body during periods of fasting, it causes severe hypoglycemia and results in increased glycogen storage in liver and kidneys. Both organs function normally in childhood, but are susceptible to a variety of problems in adult years. Other metabolic derangements include lactic acidosis and hyperlipidemia. Frequent or continuous feedings of cornstarch or other carbohydrates are the principal treatment. Other therapeutic measures may be needed for associated problems.
The disease was named after German doctor Edgar von Gierke.[3][4]
Genetics and molecular biology
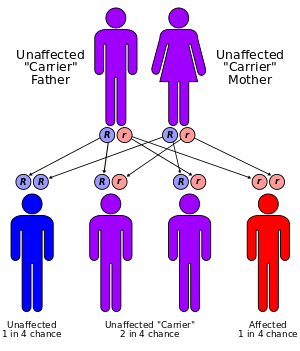
GSD Ia is inherited as an autosomal recessive disease. Heterozygote carriers are asymptomatic. As for other autosomal recessive diseases, the recurrence risk for each subsequent child of the same parents is 25%. Prenatal diagnosis has been made by fetal liver biopsy at 18–22 weeks of gestation, but no fetal treatment has been proposed. Prenatal diagnosis is possible with fetal DNA obtained by chorionic villus sampling when a fetus is known to be at risk.
Glucose-6-phosphatase is an enzyme located on the inner membrane of the endoplasmic reticulum. The catalytic unit is associated with a calcium binding protein, and three transport proteins (T1, T2, T3) that facilitate movement of glucose-6-phosphate (G6P), phosphate, and glucose (respectively) into and out of the enzyme.
The most common forms of GSD I are designated GSD Ia and GSD Ib, the former accounting for over 80% of diagnosed cases and the latter for less than 20%. A few rarer forms have been described.
- GSD Ia results from mutations of G6PC, the gene for glucose-6-phosphatase,[5] located on chromosome 17q21.[6]
- GSD Ib results from mutations of the gene for SLC37A4 or "G6PT1", the G6P transporter.[6][7]
- GSD Ic results from mutations of SLC17A3 or SLC37A4.[8]
The metabolic characteristics of GSD Ia and Ib are quite similar, but Ib incurs a few additional problems (described below).
Metabolic pathophysiology
Normal carbohydrate balance and maintenance of blood glucose levels
Glycogen in liver and (to a lesser degree) kidneys serves as a form of stored, rapidly accessible glucose, so that the blood glucose level can be maintained between meals. For about 3 hours after a carbohydrate-containing meal, high insulin levels direct liver cells to take glucose from the blood, to convert it to glucose-6-phosphate (G6P) with the enzyme glucokinase, and to add the G6P molecules to the ends of chains of glycogen (glycogen synthesis). Excess G6P is also shunted into production of triglycerides and exported for storage in adipose tissue as fat.
When digestion of a meal is complete, insulin levels fall, and enzyme systems in the liver cells begin to remove glucose molecules from strands of glycogen in the form of G6P. This process is termed glycogenolysis. The G6P remains within the liver cell unless the phosphate is cleaved by glucose-6-phosphatase. This dephosphorylation reaction produces free glucose and free PO
4 anions. The free glucose molecules can be transported out of the liver cells into the blood to maintain an adequate supply of glucose to the brain and other organs of the body. Glycogenolysis can supply the glucose needs of an adult body for 12–18 hours.
When fasting continues for more than a few hours, falling insulin levels permit catabolism of muscle protein and triglycerides from adipose tissue. The products of these processes are amino acids (mainly alanine), free fatty acids, and lactic acid. Free fatty acids from triglycerides are converted to ketones, and to acetyl-CoA. Amino acids and lactic acid are used to synthesize new G6P in liver cells by the process of gluconeogenesis. The last step of normal gluconeogenesis, like the last step of glycogenolysis, is the dephosphorylation of G6P by glucose-6-phosphatase to free glucose and PO
4.
Thus glucose-6-phosphatase mediates the final, key, step in both of the two main processes of glucose production during fasting. In fact the effect is amplified because the resulting high levels of glucose-6-phosphate inhibit earlier key steps in both glycogenolysis and gluconeogenesis.
Pathophysiology
The principal metabolic effects of deficiency of glucose-6-phosphatase are: hypoglycemia, lactic acidosis, hypertriglyceridemia, and hyperuricemia.
The hypoglycemia of GSD I is termed "fasting", or "post-absorptive", usually about 4 hours after the complete digestion of a meal. This inability to maintain adequate blood glucose levels during fasting results from the combined impairment of both glycogenolysis and gluconeogenesis. Fasting hypoglycemia is often the most significant problem in GSD I, and typically the problem that leads to the diagnosis. Chronic hypoglycemia produces secondary metabolic adaptations, including chronically low insulin levels and high levels of glucagon and cortisol.
Lactic acidosis arises from impairment of gluconeogenesis. Lactic acid is generated both in the liver and muscle and is oxidized by NAD+ to pyruvic acid and then converted via the gluconeogenic pathway to G6P. Accumulation of G6P inhibits conversion of lactate to pyruvate. The lactic acid level rises during fasting as glucose falls. In people with GSD I, it may not fall entirely to normal even when normal glucose levels are restored.
Hypertriglyceridemia resulting from amplified triglyceride production is another indirect effect of impaired gluconeogenesis, amplified by chronically low insulin levels. During fasting, the normal conversion of triglycerides to free fatty acids, ketones, and ultimately acetyl-CoA is impaired. Triglyceride levels in GSD I can reach several times normal and serve as a clinical index of "metabolic control".
Hyperuricemia results from a combination of increased generation and decreased excretion of uric acid, which is generated when increased amounts of G6P are metabolized via the pentose phosphate pathway. It is also a byproduct of purine degradation. Uric acid competes with lactic acid and other organic acids for renal excretion in the urine. In GSD I increased availability of G6P for the pentose phosphate pathway, increased rates of catabolism, and diminished urinary excretion due to high levels of lactic acid all combine to produce uric acid levels several times normal. Although hyperuricemia is asymptomatic for years, kidney and joint damage gradually accrue.
Principal clinical problems
Clinical manifestations result, directly or indirectly, from
- inability to maintain an adequate blood glucose level during the post-absorptive hours of each day
- organ changes due to glycogen accumulation
- excessive lactic acid generation
- damage to tissue from hyperuricemia
- in GSD Ib, bleeding and infection risk from blood cell effects
Hypoglycemia
Hypoglycemia is the central clinical problem, the one that is most damaging, and the one that most often prompts the initial diagnosis. Maternal glucose transferred across the placenta prevents hypoglycemia in a fetus with GSD I, but the liver is enlarged with glycogen at birth. The inability to generate and release glucose soon results in hypoglycemia, and occasionally in lactic acidosis fulminant enough to appear as a primary respiratory problem in the newborn period. Neurological manifestations are less severe than if the hypoglycemia were more acute. The brain's habituation to mild hypoglycemia is at least partly explained by use of alternative fuels, primarily lactate.
More commonly, infants with GSD I tolerate without obvious symptoms a chronic, mild hypoglycemia, and compensated lactic acidosis between feedings. Blood glucose levels are typically 25 to 50 mg/dl (1.4–2.8 mM). These infants continue to need oral carbohydrates every few hours. Many never sleep through the night even in the second year of life. They may be pale, clammy, and irritable a few hours after a meal. Developmental delay is not an intrinsic or inevitable effect of glucose-6-phosphatase deficiency but is common if the diagnosis is not made in early infancy.
Although mild hypoglycemia for much of the day may go unsuspected, the metabolic adaptations described above make severe hypoglycemic episodes, with unconsciousness or seizure, uncommon before treatment. Episodes which occur are likely to happen in the morning before breakfast. GSD I is therefore a potential cause of ketotic hypoglycemia in young children.
Once the diagnosis has been made, the principal goal of treatment is to maintain an adequate glucose level and prevent hypoglycemia.
Hepatomegaly and liver problems
Impairment of glycogenolysis also causes the characteristic enlargement of the liver due to accumulation of glycogen. Glycogen also accumulates in kidneys and small intestine. Hepatomegaly, usually without splenomegaly, begins to develop in fetal life and is usually noticeable in the first few months of life. By the time the child is standing and walking, the hepatomegaly may be severe enough to cause the abdomen to protrude. The liver edge is often at or below the level of the umbilicus. Other liver functions are usually spared, and liver enzymes and bilirubin are usually normal.
Glucose-6-phosphatase deficiency increases the risk of hepatic adenoma. There is some evidence that metabolic control of the disease is a factor.[9]
Lactic acidosis
Impaired gluconeogenesis results in elevations of lactic acid (4–10 mM) even when the child is well. In an episode of metabolic decompensation, lactic acid levels abruptly rise and can exceed 15 mM, producing severe metabolic acidosis. Uric acid, ketoacids, and free fatty acids further increase the anion gap. Manifestations of severe metabolic acidosis include vomiting and hyperpnea, which can exacerbate hypoglycemia by reducing oral intake. Repeated episodes of vomiting with hypoglycemia and dehydration may occur in infancy and childhood, precipitated by (or mimicking) infections such as gastroenteritis or pneumonia.
Growth failure
Without treatment, growth failure is common, due to chronically low insulin levels, persistent acidosis, chronic elevation of catabolic hormones, calorie insufficiency, and/or malabsorption.
Hyperlipidemia and blood vessel effects
A secondary effect of low insulin levels is hypertriglyceridemia. Triglycerides in the 400–800 mg/dl range may produce visible lipemia, and even a mild pseudohyponatremia due to a reduced aqueous fraction of the serum. Cholesterol is only mildly elevated. Immobilisation of fats results in an increase in Fatty Acids and ketone bodies.
Hyperuricemia and joint problems
A further effect of chronic lactic acidosis in GSD I is hyperuricemia, as lactic acid and uric acid compete for the same renal tubular transport mechanism. Increased purine catabolism is an additional contributing factor. Uric acid levels of 6–12 mg/dl are typical of GSD I. Allopurinol may be needed to prevent uric acid nephropathy and gout.
Kidney effects
Kidneys are usually 10 to 20% enlarged with stored glycogen. This does not usually cause clinical problems in childhood, with the occasional exception of a Fanconi syndrome with multiple derangements of renal tubular reabsorption, including proximal renal tubular acidosis with bicarbonate and phosphate wasting. However, prolonged hyperuricemia can cause uric acid nephropathy. In adults with GSD I, chronic glomerular damage similar to diabetic nephropathy may lead to renal failure.
Bowel effects
Intestinal involvement can cause mild malabsorption with steatorrhea, greasy stools, but usually requires no treatment.
Infection risk
Neutropenia is a manifestation of this disease. Granulocyte colony-stimulating factor (G-CSF, e.g. filgrastim) therapy can reduce the risk of infection.
Blood clotting problems
Impaired platelet aggregation is an uncommon effect of chronic hypoglycemia. It may cause clinically significant bleeding, especially epistaxis.
Neurodevelopmental effects
Developmental delay is a potential secondary effect of chronic or recurrent hypoglycemia, but is at least theoretically preventable. Normal neuronal and muscle cells do not express glucose-6-phosphatase, so GSD I causes no other neuromuscular effects.
Presentation and diagnosis
Several different problems may lead to the diagnosis, usually by two years of age:
- seizures or other manifestations of severe fasting hypoglycemia
- hepatomegaly with abdominal protuberance
- hyperventilation and apparent respiratory distress due to metabolic acidosis
- episodes of vomiting due to metabolic acidosis, often precipitated by minor illness and accompanied by hypoglycemia
Once the diagnosis is suspected, the multiplicity of clinical and laboratory features usually makes a strong circumstantial case. If hepatomegaly, fasting hypoglycemia, and poor growth are accompanied by lactic acidosis, hyperuricemia, hypertriglyceridemia, and enlarged kidneys by ultrasound, gsd I is the most likely diagnosis. The differential diagnosis list includes glycogenoses types III and VI, fructose 1,6-bisphosphatase deficiency, and a few other conditions (page 5), but none are likely to produce all of the features of GSD I.
The next step is usually a carefully monitored fast. Hypoglycemia often occurs within six hours. A critical blood specimen obtained at the time of hypoglycemia typically reveals a mild metabolic acidosis, high free fatty acids and beta-hydroxybutyrate, very low insulin levels, and high levels of glucagon, cortisol, and growth hormone. Administration of intramuscular or intravenous glucagon (0.25 to 1 mg, depending on age) or epinephrine produces little rise of blood sugar.
The diagnosis is definitively confirmed by liver biopsy with electron microscopy and assay of glucose-6-phosphatase activity in the tissue and/or specific gene testing, available in recent years.
Treatment
The primary treatment goal is prevention of hypoglycemia and the secondary metabolic derangements by frequent feedings of foods high in glucose or starch (which is readily digested to glucose). To compensate for the inability of the liver to provide sugar, the total amount of dietary carbohydrate should approximate the 24-hour glucose production rate. The diet should contain approximately 65–70% carbohydrate, 10–15% protein, and 20–25% fat. At least a third of the carbohydrates should be supplied through the night, so that a young child goes no more than 3–4 hours without carbohydrate intake
In the last 30 years, two methods have been used to achieve this goal in young children: (1) continuous nocturnal gastric infusion of glucose or starch; and (2) night-time feedings of uncooked cornstarch. An elemental formula, glucose polymer, and/or cornstarch can be infused continuously through the night at a rate supplying 0.5–0.6 g/kg/h of glucose for an infant, or 0.3–0.4 for an older child. This method requires a nasogastric or gastrostomy tube and pump. Sudden death from hypoglycemia has occurred due to malfunction or disconnection, and periodic cornstarch feedings are now preferred to continuous infusion.
Cornstarch is an inexpensive way to provide gradually digested glucose. One tablespoon contains nearly 9 g carbohydrate (36 calories). Although it is safer, less expensive, and requires no equipment, this method does require that parents arise every 3–4 hours to administer the cornstarch. A typical requirement for a young child is 1.6 g/kg every 4 hours.
Long-term management should eliminate hypoglycemic symptoms and maintain normal growth. Treatment should achieve normal glucose, lactic acid, and electrolyte levels, and only mild elevations of uric acid and triglycerides.
Avoidance of other sugars
Intake of carbohydrates which must be converted to G6P to be utilized (e.g., galactose and fructose) should be minimized. Although elemental formulas are available for infants, many foods contain fructose or galactose in the forms of sucrose or lactose. Adherence becomes a contentious treatment issue after infancy.
Other therapeutic measures
Persistent elevation of uric acid above 6.5 mg/dl warrants treatment with allopurinol to prevent uric acid deposition in kidneys and joints.
Because of the potential for impaired platelet function, coagulation ability should be checked and the metabolic state normalized before surgery. Bleeding time may be normalized with 1–2 days of glucose loading, and improved with ddavp. During surgery, iv fluids should contain 10% dextrose and no lactate.
A patient with GSD, type 1b was treated with a liver transplant at UCSF Medical Center in 1993 that resulted in the resolution of hypoglycemic episodes and the need for the patient to stay away from natural sources of sugar. Other patients have undergone this procedure as well with positive results. Although a liver transplant resulted in the resolution of hypoglycemia it did not however resolve the chronic neutropenia and the risk of infection among patients.
Treatment of acute metabolic acidosis episodes
The most significant acute problem in childhood is a vulnerability to episodes of metabolic acidosis precipitated by minor illnesses. If a vomiting illness persists longer than 2–4 hours, the child should be seen and assessed for dehydration, acidosis, and hypoglycemia. If these are developing, intravenous fluids should be provided at a rate above maintenance. For mild acidosis, an effective fluid is 10% dextrose in ½ normal saline with 20 mEq/l KCl, but if acidosis is severe, 75–100 mEq/l NaHCO
3 and 20 mEq/l of K acetate can be substituted for the NaCl and KCl.
Natural history, prognosis, long term complications
Without adequate metabolic treatment, patients with gsd I have died in infancy or childhood of overwhelming hypoglycemia and acidosis. Those who survived were stunted in physical growth and delayed in puberty because of chronically low insulin levels. Mental retardation from recurrent, severe hypoglycemia is considered preventable with appropriate treatment.
Hepatic complications have been serious in some patients. Adenomas of the liver can develop in the second decade or later, with a small chance of later malignant transformation to hepatoma or hepatic carcinomas (detectable by alpha-fetoprotein screening). Several children with advanced hepatic complications have improved after liver transplantation.
Additional problems reported in adolescents and adults with gsd I have included hyperuricemic gout, pancreatitis, and chronic renal failure. Despite hyperlipidemia, atherosclerotic complications are uncommon.
With diagnosis before serious harm occurs, prompt reversal of acidotic episodes, and appropriate long-term treatment, most children will be healthy. With exceptions and qualifications, adult health and life span may also be fairly good, although lack of effective treatment before the mid-1970s has limited our long-term information.
Epidemiology
In the United States, GSD Ia has an incidence of approximately 1 in 50,000[1] to 100,000[2] births. None of the glycogenoses are currently detected by standard or extended newborn screening.
The disease is more common in Ashkenazi Jewish populations, people of Mexican, Chinese, and Japanese descent.[10]
References
- 1 2 Glycogen-Storage Disease Type I at eMedicine
- 1 2 The Association for Glycogen Storage Disease: Type I Glycogen Storage Disease Type I GSD This page was created in October 2006.
- ↑ Gierke's syndrome at Who Named It?
- ↑ von Gierke, E. (1929). "Hepato-nephromegalia glykogenica (Glykogenspeicherkrankheit der Leber und Nieren)". Beiträge zur pathologischen Anatomie und zur allgemeinen Pathologie. Jena. 82: 497–513.
- ↑ "Glycogen storage disease type I". Genetics Home Reference from U.S. National Library of Medicine & National Institutes of Health. Retrieved 6 July 2013.
- 1 2 Bali, DS; Chen, YT; Goldstein, JL; Pagon, RA; Adam, MP; Bird, TD; Dolan, CR; Fong, CT; et al. (1993). "Glycogen Storage Disease Type I". PMID 20301489.
- ↑ Online Mendelian Inheritance in Man (OMIM) Glycogen Storage Disease Ib -232220
- ↑ Online Mendelian Inheritance in Man (OMIM) Glycogen Storage Disease Ic -232240
- ↑ Wang, DQ; Fiske, LM; Carreras, CT; Weinstein, DA (September 2011). "Natural history of hepatocellular adenoma formation in glycogen storage disease type I.". The Journal of Pediatrics. 159 (3): 442–6. doi:10.1016/j.jpeds.2011.02.031. PMC 3135733
 . PMID 21481415.
. PMID 21481415. - ↑ Goldman, Lee (2011). Goldman's Cecil Medicine (24th ed.). Philadelphia: Elsevier Saunders. p. 1356. ISBN 1437727883.
External links
- GeneReview/NIH/UW entry on Glycogen Storage Disease Type I
- Asociación Española de Enfermos de Glucogenosis