Alpha-mannosidosis
| Alpha-mannosidosis | |
|---|---|
| Classification and external resources | |
| Specialty | endocrinology |
| ICD-10 | E77.1 |
| ICD-9-CM | 271 |
| OMIM | 248500 |
| DiseasesDB | 31422 |
| MeSH | D008363 |
| GeneReviews | |
Alpha-mannosidosis is a lysosomal storage disorder[1] caused by deficient activity of the enzyme alpha-D-mannosidase. In humans it is known to be caused by an autosomal recessive genetic mutation.[2] In livestock it is caused by chronic poisoning with swainsonine from locoweed.
Pathophysiology

A defective alpha-mannosidase enzyme, which normally helps to break down complex sugars derived from glycoproteins in the lysosome, causes sugar build up and impairs cell function. Complete absence of functionality in this enzyme leads to death during early childhood due to deterioration of the central nervous system. Enzymes with low residual activity lead to a milder type of the disease, with symptoms like reduced hearing, mental disabilities, susceptibility to bacterial infections, and skeletal deformities. The course of the disease is progressive.
Alpha-mannosidosis is classified into types I through III based on severity and age of onset. In contrast to the usual classifications scheme of these disorders, type III is the most severe.
Symptoms

Symptoms range widely in their onset and severity. The onset of the most severe form, type III, begins within the first months of life and includes a quick progression of intellectual disability, liver and spleen enlargement (splenomegaly), hearing loss, respiratory infections and skeletal abnormalities. Often the appearance of an affected individual includes the following facial features: protruding forehead, leveled nasal bridge, small nose and wide mouth. Muscular weakness or spinal abnormalities can occur due to the buildup of storage materials in the muscle. A milder form of alpha-mannosidosis involves mild to moderate intellectual disability which develops during childhood or adolescence.
Diagnosis and testing
A diagnosis is made by measuring the enzymatic activity of alpha-D-mannosidase in white blood cells. If there is a decreased level of the enzyme in comparison to standard levels, a diagnosis can be made. It is thought that this disorder might be under-diagnosed for a few different reasons—the diagnosis is often made late in the disease's progression, symptoms are often mild, or the biochemical diagnosis does not yield conclusive results.
Life expectancy
The life expectancy in alpha-mannosidosis is highly variable. Individuals with early onset severe disease often do not survive beyond childhood, whereas those with milder disorders may survive well into adult life.
Treatment
There is no cure for congenital alpha-mannosidosis. Treatment is limited to reducing or controlling the symptoms of this disorder by, for example, taking medication to control seizures, using a hearing aid to assist with hearing loss, and by having routine physical therapy to assist with muscular pain and weakness. In some cases, a wheelchair is recommended if muscle or spinal impairments immobilize the individual affected. Despite early reports to the contrary,[3] bone marrow transplants performed at an early age have shown promise in halting the progression of this disorder.[4]
Genetic prevalence
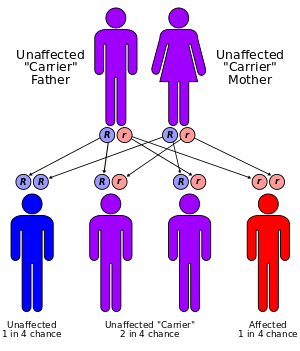
The worldwide incidence of alpha-mannosidosis is in the range of 1 per 500,000[4] to 1 per 1,000,000. Mannosidosis is found in all ethnic groups in Europe, America, Africa, and Asia.
References
- ↑ Roces DP, Lüllmann-Rauch R, Peng J, et al. (2004). "Efficacy of enzyme replacement therapy in alpha-mannosidosis mice: a preclinical animal study". Hum. Mol. Genet. 13 (18): 1979–88. doi:10.1093/hmg/ddh220. PMID 15269179.
- ↑ Gotoda Y, Wakamatsu N, Kawai H, Nishida Y, Matsumoto T (October 1998). "Missense and nonsense mutations in the lysosomal alpha-mannosidase gene (MANB) in severe and mild forms of alpha-mannosidosis". American Journal of Human Genetics. 63 (4): 1015–24. doi:10.1086/302048. PMC 1377481
 . PMID 9758606.
. PMID 9758606. - ↑ Will A, et al. (1987). "Bone marrow transplantation in the treatment of alpha-mannosidosis". Disease in Childhood. 62 (10): 1044–1049. doi:10.1136/adc.62.10.1044.
- 1 2 Malm D, Nilssen O (2008). "Alpha-mannosidosis". Orphanet J Rare Dis. 3 (1): 21. doi:10.1186/1750-1172-3-21. PMC 2515294. PMID 18651971.
External links
- GeneReviews/NCBI/NIH/UW entry on Alpha-Mannosidosis
- OMIM entries on Alpha-Mannosidosis
- ISMRD page on alpha-mannosidosis
- Hide and Seek Foundation For Lysosomal Disease Research
- Alpha-mannosidosis at DMOZ
- Alpha-mannosidosis type 1 at NIH's Office of Rare Diseases
- Alpha-mannosidosis type 2 at NIH's Office of Rare Diseases